Overall Objectives
Introduction
Constraint Logic Programming supports a great ambition for
programming: the one of making of programming essentially a
modeling task, with equations, constraints and logical
formulas. This research field is born during the mid 80s from
Logic Programming, Linear Programming coming from Operations
Research, and Constraint Propagation techniques coming from
Artificial Intelligence. Its foundation is the use of
relations on mathematical variables to compute with partial
information.
The successes of Constraint Programming for solving
combinatorial optimization problems, ranging from pure
academic problems to real-life problems in industry or
commerce, owe much to the bringing of both new local
consistency techniques and new declarative languages which
allow control on the mixing of heterogeneous resolution
techniques: numerical, symbolic, deductive and heuristic.
Furthermore, this approach of combinatorial problems based
on logical modeling can be fruitfully applied in systems
biology to get over complexity walls and reason about large
molecular interaction networks.
The "Contraintes" group investigates the logical
foundations, design, implementation, programming environments
and applications of constraint languages. The study of
Concurrent Constraint languages is a core aspect of the
project as they provide a conceptual framework for analyzing
different issues of constraint programming, like constraint
resolution techniques, concurrent modeling, reactive
applications, etc. The main application domains investigated
are combinatorial optimization problems and computational
systems biology. In bioinformatics, our objective is not to
work on structural biology problems which has been the main
trend up to now, but to attack the great challenge of systems
biology, namely to model the function, activity and
interaction of molecular processes in living cells, with
logic programming concepts and program verification
techniques.
Highlight: Model Reductions as Subgraph
Epimorphisms
In Systems Biology, an increasing collection of reaction
rule based models of various biological processes is
currently developed and made available in publicly accessible
repositories, such as biomodels.net for instance (currently
composed of 269 curated models), using common exchange
formats such as the Systems Biology Markup Language (SBML).
To date, however, there is no general method to relate
different models to each other by abstraction or reduction
relationships, and this task is left to the modeler for
re-using and coupling models. In mathematical biology, model
reduction techniques have been studied for a long time,
mainly in the case where a model exhibits different time
scales, or different spatial phases, which can be analyzed
separately. These techniques are however far too restrictive
to be applied on a large scale in systems biology, and do not
take into account abstractions other than time or phase
decompositions.
In
Subgraph epimorphisms are also interesting combinatorial
objects that we now study in their own rights. In
collaboration with Christine Solnon (INSA Lyon), we develop
their theory and compare them with the classical notions of
subgraph isomorphisms, minors, etc.
Scientific Foundations
Concurrent Constraint Logic
Programming
The class of Concurrent Constraint programming languages
(CC) was introduced two decades ago by Vijay Saraswat as a
unifying framework for constraint logic programming and
concurrent logic programming. The CC paradigm constitutes a
representative abstraction of practical constraint
programming languages, allowing for a theoretical study of
their fundamental properties.
CC extends the Constraint Logic Programming framework
(CLP) by introducing a synchronization primitive, based on
constraint entailment. This provides a model of concurrent
computation, where agents communicate through a shared store,
represented by a constraint, which expresses some
partial informationon the values of the variables
involved in the computation. The variables play the role of
transmissible dynamically created communication channels.
One important success of CC has been the simple and
elegant reconstruction of finite domain constraint solvers,
and the cooperation of several models to solve a single
combinatorial problem. The programming of reactive
applications however motivates new extensions, in particular
for relaxing the hypothesis of monotonic evolution of the
constraint store and expressing state change.
One solution to this problem was found by considering the
logical semantics of CC languages. The completeness theorems
which relate the execution of a CLP program to its
translation in classical logic are indeed broken by the
synchronization operation of CC. Looking for a logical
semantics of CC programs in the general paradigm of logic
programming,
program = logical formula,
execution = proof search,
leads to a translation in Jean-Yves
Girard's linear logic, in which the CC operational
transitions are identified to linear implications (in the
opposite direction of the classical CLP logical semantics).
The associated completeness theorems for successes and
accessible stores (and suspensions in the non-commutative
logic of Ruet and Abrusci) continue to hold for linear logic
constraint systems which allow for non monotonic evolution of
the constraint store.
In this theoretical framework, called LCC for Linear Logic
Concurrent Constraint programming
F. Fages, P. Ruet, S.
Soliman.
Linear concurrent constraint programming: operational and
phase semantics, in “Information and Control”, 2001,
vol. 165(1), pp.14-41., it is thus possible
to address important issues for Constraint Programming:
verifying CC programs;
combining CC and state-based
programming;
defining extensible libraries of
constraint solvers without changing of programming
language.
Constraint Filtering Algorithms
The project works on constraint resolution methods and
their cooperation. The main focus is the declarativeness of
the constraint solver, the efficiency of constraint
propagation methods, the design of global constraints and the
combination of constraint propagation with heuristic
search.
Our application domains involve quite different constraint
domains on which we use or contribute with new constraint
filtering algorithms for
finite domains (bounded natural
numbers): membership, arithmetic, reified, higher order
and global constraints;
reals: polyhedral libraries for linear
constraints and interval methods otherwise;
terms: subtyping constraints;
graphs: subgraph epimorphism and
isomorphism constraints;
Kripke structures: temporal logic
constraints (QFCTL and QFLTL formulae with free
variables).
Logical Paradigm for Systems Biology
At the end of the Nineties, research in Bioinformatics
evolved, passing from the analysis of the genomic sequence to
the analysis of post-genomic interaction networks (expression
of RNA and proteins, protein-protein interactions, etc). The
complexity of these networks requires a large research effort
to develop symbolic notation and analysis tools for
biological processes and data. In order to scale-up, and get
over the complexity walls to reason about biological systems,
there is a general feeling that beyond providing tools to
biologists, computer science has much to offer in terms of
concepts and methods. Systems biology is the name given to a
pluridisciplinary domain involving biology, computer science,
mathematics, physics,... and aiming at elucidating the
high-level functions of the living organisms from their
biochemical bases at the molecular level.
Our main research axis in this field since 2002 has been
the application of logic programming concepts and program
verification techniques to the modeling and analysis of
complex biochemical processes in the cell. The logical
paradigm for systems biology that we develop can be
summarized by the following identifications :
biological model = (quantitative) state transition
system,
biological property = temporal logic formula,
model validation = model-checking,
model inference = constraint solving.
This original approach can combine different levels of
abstraction, both qualitative and quantitative, for reasoning
about cell processes. It has proven successful for the
modeling, analysis and even optimization of complex molecular
processes in the cell.
Application Domains
Combinatorial optimization problems
The number and economic impact of combinatorial
optimization problems found in the industrial world are
constantly increasing. They cover:
resource allocation;
placement, bin packing;
scheduling;
planning;
transport;
etc.
The last forty years have brought many improvements in
Operations Research resolution techniques. In this context,
Constraint Programming can be seen as providing, on the one
hand, local consistency techniques that can be applied to
various numerical or symbolic constraints, and on the other
hand, declarative languages to model real-life problems and
express complex resolution strategies. The latter point is
crucial for designing new algorithms that cannot be defined
without a sufficiently high-level language to express them.
It allowed for better results than traditional methods, for
instance in scheduling, and is promised to an even better
future when thinking about the cooperation of global
resolution, local consistency techniques and search
methods.
The project builds upon its knowledge of constraint
languages, constraint solvers and their implementation to
work in these directions. The LCC paradigm offers at the same
time a theoretical framework for analysis, and a valuable
guide for practical language design and implementation. The
work on programming environments helps to integrate the
Constraint Programming tools into this application
domain.
The European FP6 Strep project
[Net-WMS]that we have
coordinated, has shown the benefit of combining discrete
geometry constraints with rules to express physical, common
sense and packing business constraints to solve packing
problems in the context of warehouse management systems for
the automotive industry
[Rules2CP], to express
requirements in a declarative and flexible manner. and
compile them to efficient constraint programs using reified
constraints and a global constraint dedicated to geometrical
placement problems in high dimension.
Computational Systems Biology
In 2002 , we started a Collaborative Research Initiative
ARC CPBIO on “Process Calculi and Biology of Molecular
Networks”. By working on well understood biological models,
we sought:
to identify in the family of
competitive models coming from the Theory of Concurrency
and from Logic Programming (Constraint Logic Programming,
Concurrent Constraint languages and their extensions to
discrete and continuous time, TCC, HCC), the ingredients
of a language for the modular and multi-scale
representation of biological processes;
to provide a series of examples of
biomolecular processes transcribed in formal languages,
and a set of biological questions of interest about these
models;
to design and apply to these examples
formal computational reasoning tools for the simulation,
the analysis and the querying of the models.
This work lead us to the design and implementation of the
Biochemical Abstract Machine
[BIOCHAM]that has the unique
feature of providing formal languages corresponding to
different qualitative and quantitative levels of abstraction
for, on the one hand, modeling biomolecular interaction
diagrams with reaction rules, and on the other hand, modeling
the biological properties of the system in temporal logic.
This double formalization of both the model and the
biological properties of the system at hand has opened
several new research avenues on the design and systematic
validation of biological models.
In partnership with biologists, we develop and experiment
our modeling technology in three domains:
Modeling of cell cycle and optimization of cancer
therapies.This research initiated in 2004 in
partnership with Jean Clairambault, EPI BANG, and Francis
Lévi INSERM, Hopital Paul Brousse, Villejuif, aims at
understanding fundamental mechanisms involved in cancer
and chronotherapies through mathematical modeling.
Following, the EU STREP project
[
TEMPO](2006-2009) on “temporal genomics for patient
tailored chronotherapeutics”, coordinated by Francis
Lévi, our development in
[
BIOCHAM]of original coupled models of the cell
cycle, the circadian clock, the DNA repair system,
irinotecan metabolism and drug injection optimization,
continues in the Era-Net SysBio
[
C5Sys]project (2010-2013) coordinated by Francis
Lévi and David Rand, University of Warwick, UK, by
focussing on the interactions between the cell cycle and
the circadian clock in mammalian cells.
Modeling and analysis of G-protein coupled receptor
signal transduction. This research initiated in 2004
in partnership with Frédérique Clément, EPI SISYPHE, and
Eric Reiter, INRA Tours, aims at understanding the
structure and the dynamics of FSH and angiotensine signal
transduction in mammalian cells. Our article submitted to
Science Signaling
[
INSIGHT](2006-2009) and in the AE
[
REGATE].
Control of gene expression and synthetic
biology.This research lead in the team by Grégory
Batt investigates the possibilities to control and
reprogram gene expression in living cells. In
collaboration with Pascal Hersen and Samuel Bottani,
biophysicists at the Matière and Systèmes Complexes lab,
CNRS/Paris Diderot University, we design a microfluidic
platform and develop control software for the real-time
control of gene expression in yeast. In the AE COLAGE
coordinated by Huges Berry, EPI Combining, with François
Taddei, Ariel Lindner, INSERM Paris Necker, Hidde de
Jong, Delphine Ropers, EPI IBIS, Jean-Luc Gouzé, and
Madalena Chaves, EPI COMORE, we investigate similarly the
possibilities to control and reprogram growth and aging
in bacteria
E. coliusing synthetic biology approaches.
Finally, in collaboration with Ron Weiss at MIT, USA, we
have started a project on the synthetic control of the
population density in a mammalian cellular tissue.
Software
BIOCHAM
François
Fages
Dragana
Jovanovska
Aurélien
Rizk
Sylvain
Soliman
The Biochemical Abstract Machine
[BIOCHAM]
[SBML]) and is interpreted with
three semantics correspnding to three abstraction levels:
the boolean semantics (presence or
absence of molecules),
the differential semantics
(concentrations of molecules),
the stochastic semantics (discrete
numbers of molecules).
Based on this formal framework, BIOCHAM features:
Boolean and numerical simulators
(Rosenbrock's method for the differential semantics,
Gillespie's algorithm with tau lipping for the stochastic
semantics);
a temporal logic language (CTL for
qualitative models and QFLTL(R) with numerical
constraints for quantitative models) for formalizing
biological properties such as reachability, checkpoints,
oscillations or stability, and checking them
automatically with model-checking techniques;
automatic search procedures to infer
parameter values, initial conditions and even reaction
rules from temporal logic properties;
automatic detection of invariants,
through constraint-based analysis of the underlying Petri
net;
an SBGN-compatible reaction graph
editor
BIOCHAM is implemented in GNU-Prolog and interfaced to the
symbolic model checker
[NuSMV]and to the continuous
optimization tool
[CMAES]developed by the EPI
TAO.
Rules2CP
François
Fages
Julien
Martin
Thierry
Martinez
[Rules2CP]is a rule-based
modeling language for constraint programming. It is
distributed since 2009 as open-source. Unlike other modeling
languages for constraint programming, Rules2CP adopts a
single knowledge representation paradigm based on rules
without recursion, and a restricted set of data structures
based on records and enumerated lists given with iterators.
This allows us to model complex constraint satisfaction
problems together with search strategies, where search trees
are expressed by logical formulae and heuristic choice
criteria are defined with preference orderings by
pattern-matching on the rules' left-hand sides.
The expressiveness of Rules2CP has been illustrated in the
FP6 Strep project
[Net-WMS]by a complete library
for packing problems, called PKML (Packing Knowledge Modeling
Library), which, in addition to pure bin packing and bin
design problems, can deal with common sense rules about
weights, stability, as well as specific packing business
rules.
SiLCC
Thierry
Martinez
[SiLCC]is an extensible modular
concurrent constraint programming language relying upon
linear logic. It is a complete implementation of the Linear
logic Concurrent Constraint programming paradigm of Saraswat
and Lincoln using the formal semantics of Fages, Ruet and
Soliman. It is a single-paradigm logical language, enjoying
concurrency, imperative traits, and a clean module system
allowing to develop hierarchies of constraint systems within
the language.
This software prototype is used to study the design of
hierarchies of extensible libraries of constraint solvers.
SiLCC is also considered as a possible implementation
language for restructuring the code of
[BIOCHAM].
CHRat
Thierry
Martinez
[CHRat]is a modular version of
the well known Constraint Handling Rules language CHR, called
for CHRat for CHR with
askand
tell. Inspired by the LCC framework, this extension of
CHR makes it possible to reuse CHRat components both in rules
and guards in other CHRat components, and define hierarchies
of constraint solvers. CHRat is a bootstrapped preprocessor
for CHR implemented in Prolog.
CLPGUI
François
Fages
[CLPGUI]is a generic graphical
user interface written in Java for constraint logic
programming. It is available for GNU-Prolog and SICStus
Prolog. CLPGUI has been developed both for teaching purposes
and for debugging complex programs. The graphical user
interface is composed of several windows: one main console
and several dynamic 2D and 3D viewers of the search tree and
of finite domain variables. With CLPGUI it is possible to
execute incrementally any goal, backtrack or recompute any
state represented as a node in the search tree. The level of
granularity for displaying the search tree is defined by
annotations in the CLP program.
CLPGUI has been mainly developped in 2001 and is
distributed as third-party software on GNU-Prolog and SICStus
Prolog web sites. In 2009, CLPGUI has been interfaced to
Rules2CP/PKML and used in FP6 Strep
[Net-WMS]with a non-released
version.
New Results
Linear Logic Concurrent Constraint Programming and
Constraint Handling Rules
François
Fages
Thierry
Martinez
Sylvain
Soliman
The Constraint Handling Rules (CHR) language of Thom
Frühwirth shares many similarities with
[SiLCC]
[CHRat]) and SiLCC are based on
a similar model of concurrent computation, where agents
communicate through a shared constraint store, with a
synchronization mechanism based on constraint entailment. In
particular, the Constraint Simplification Rules (CSR) subset
of CHR and the flat subset of LCC, where agent nesting is
restricted, are very close syntactically and
semantically.
In
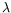 -lifting in functional languages. In conjunction with
the equivalence between CHR and CSR with respect to naive
operational semantics, these results lead to
semantics-preserving translations from full LCC to CHR and
conversely. Immediate consequences of this work include new
proofs for CHR linear logic and phase semantics, relying on
corresponding results for LCC, plus an encoding of the
-calculus in CHR.
-lifting in functional languages. In conjunction with
the equivalence between CHR and CSR with respect to naive
operational semantics, these results lead to
semantics-preserving translations from full LCC to CHR and
conversely. Immediate consequences of this work include new
proofs for CHR linear logic and phase semantics, relying on
corresponding results for LCC, plus an encoding of the
-calculus in CHR.
All the proofs of this paper are currently developed in
the Coq proof assistant with interactions with the EPI
GALLIUM.
Rule-based Modeling for Constraint
Programming
Nicolas
Beldiceanu
François
Fages
Julien
Martin
Thierry
Martinez
[Rules2CP][Net-WMS]
One originality of
[Rules2CP]as a modeling
language is its capability to express complex search
strategies, by defining search trees with logical formulae
and heuristic choice criteria by pattern matching on rule
left-hand sides' derivation. in
Theory of Traces
Pierre
Deransart
François
Fages
We are working on a general theory of traces design taking
traces as primary objects of study. It is based on the
observation of the way trace files are accumulated as
knowledge bases and elaborated in differents fields of
activity like software engeneering, rule based systems and
resolution, learning in context, or personal experience
storing systems. The state of this work is regularly updated
on
We worked on two main points: the development of a
versatile tracer of CHR
vwith several extensions, in the framework called
CHROME-REF (see TODAS project) and an application field we
called “exo-memory”. In
Model Reductions as Subgraph
Epimorphisms
François
Fages
Steven
Gay
Sylvain
Soliman
In
In collaboration with Christine Solnon (INSA Lyon), we
develop the theory of subgraph epimorphisms and compare them
with the classical notions of subgraph isomorphisms, minors,
etc.
Steady-State Analyses and Petri Nets
François
Fages
Faten
Nabli
Sylvain
Soliman
In Systems Biology, very few analyses permit to extract
information about the dynamics of quantitative models lacking
precise parameter values, when pure symbolic computations
fails. In
[Biomodels]repository.
Some other links between Petri nets and various Systems
Biology modelling formalisms are studied in the framework of
the ANR
[CALAMAR]
Indeed, structure-related qualitative analysis techniques
have become increasingly popular in recent years, like
qualitative model checking or pathway analysis (elementary
modes, invariants, flux balance analysis, graph-based
analyses, chemical organization theory, etc.). They do not
rely on kinetic information but require a well-defined
structure as stochastic analysis techniques equally do. In
Numerical Parameter Inference from Temporal Logic
Properties
Grégory
Batt
Marco
Bottalico
Elisabetta
De Maria
François
Fages
Domitille
Heitzler
Sylvain
Pradalier
Sylvain
Soliman
Aurélien
Rizk
Jannis
Uhlendorf
In
[BIOCHAM]
[CMAES]of Nikolaus Hansen from
the EPI TAO.in order to find unknown parameter
values in a model satisfying a set of biological properties
formalized in temporal logic
Following this route, similar metrics and methods are
currently developed for stochastic processes
Consistency techniques for hybrid ODE-stochastic
simulations are also studied in this perspective
Parameter Inference for Qualitative Models of
Regulatory Networks
Grégory
Batt
Investigating the relation between the structure and
behavior of complex biological networks often involves posing
the question if the hypothesized structure of a regulatory
network is consistent with the observed behavior, or if a
proposed structure can generate a desired behavior. In
Modeling the cell cycle and optimization of cancer
chronotherapies
Elisabetta
De Maria
François
Fages
Aurélien
Rizk
Sylvain
Soliman
Denis
Thieffry
Recent advances in cancer chronotherapy techniques support
the evidence that there exist some links between the cell
cycle and the circadian clock genes. One purpose for modeling
the entrainment in period of the cell cycle by the circadian
clock is to better understand how to efficiently target
malignant cells depending on the phase of the day and patient
characterictics. This is at the heart of our participation in
the EraNet SysBio project
[C5Sys], follow up of the
former EU STREP project
[TEMPO], in collaboration with
the EPI BANG.
In
[BIOCHAM]for parameter search
(running on a cluster of 10000 processors at the GENCI) can
be used to couple dynamical models in high dimension. This
approach is illustrated by a coupled model composed of:
a four phases model of the mammalian
cell cycle by Novak and Tyson,
a circadian clock model by Leloup and
Goldbeter,
a DNA damage repair model by Ciliberto
et al.,
a model of irinotecan metabolism by
Dimitrio and Ballesta,
a simple model of drug administration
control.
This coupled model allows us to minimize the toxicity of
irinotecan on healthy cells, using BIOCHAM's parameter search
method applied on the drug administration control law.
Our technology is ready to calibrate models on real
patient data, evaluate model predictions and optimize
patient-tailored chronotherapeutics. The collaboration
currently focuses on the obtaining of consistent data in the
[C5Sys]project and on the
improvement of the cell cycle model.
Modeling and analysis of FSH and Angiotensine
Signaling Networks
François
Fages
Domitille
Heitzler
Aurélien
Rizk
Sylvain
Soliman
In collaboration with Eric Reiter (UMR CNRS-INRA 6175) and
Frédérique Clément (SISYPHE) in the framework of the
Initiative Action
[REGATE], we analyse the
connectivity and dynamics of the FSH signaling network in its
target cells, and embedding the network within a multi-scale
representation, from the molecular up to the organic
level.
In
[BIOCHAM]an abstracted
ODE-based model that captured the available knowledge and
experimental data. We inferred the unknown parameters by
simultaneously fitting experimental data generated in both
control and perturbed conditions, using a cluster of 10000
processors at the GENCI. The mathematical model predicts and
experiments confirm that, for the AT1AR expressed in HEK293
cells: i) GRK2/3 and 5/6 regulate switching between the G
protein and β-arrestin pathways as well as their distinct
dynamics by phosphorylating the C- terminal region of the
activated receptor; ii) GRK2/3 not only mediates
desensitization of G protein activation but also exerts a
strong restraining influence on β-arrestin signaling; iii)
GRK5/6 exert little effect on G protein-stimulated ERK but
are required for β-arrestin- mediated ERK activation; iv)
the β-arrestin-dependent ERK pathway undergoes both
activation and deactivation through amplified enzymatic
processes.
These results convincingly illustrate the value of using
computational modeling to decipher the complex signaling
mechanisms elicited by 7TMRs
Control of the HOG signaling cascade
Grégory
Batt
François
Fages
Jannis
Uhlendorf
To decipher the dynamical functioning of cellular
processes, the method of choice is to observe the time
response of cells subjected to well controlled perturbations
in time and amplitude. Efficient methods, based on molecular
biology, are available to monitor quantitatively and
dynamically many cellular processes. In contrast, it is still
a challenge to perturb cellular processes - such as gene
expression - in a precise and controlled manner. In
We have experimentally demonstrated the feasibility of
this approach and investigated computationally some possible
improvements of our control strategy using a model of the
yeast osmo-adaptation response fitted to our data