

Section: Research Program
Models of genome evolution
Classical artificial evolution frameworks lack the basic structure of biological genome (i.e. a double-strand sequence supporting variable size genes separated by variable size intergenic sequences). Yet, if one wants to study how a mutation-selection process is likely (or not) to result in particular biological structures, it is mandatory that the effect of mutation modifies this structure in a realistic way. We have developed an artificial chemistry based on a mathematical formulation of proteins and of the phenotypic traits. In our framework, the digital genome has a structure similar to prokaryotic genomes and a non-trivial genotype-phenotype map. It is a double-stranded genome on which genes are identified using promoter-terminator- like and start-stop-like signal sequences. Each gene is transcribed and translated into an elementary mathematical element (a “protein”) and these elements - whatever their number - are combined to compute the phenotype of the organism. The Aevol (Artificial EVOLution) model is based on this framework and is thus able to represent genomes with variable length, gene number and order, and with a variable amount of non-coding sequences (for a complete description of the model, see [59] ).
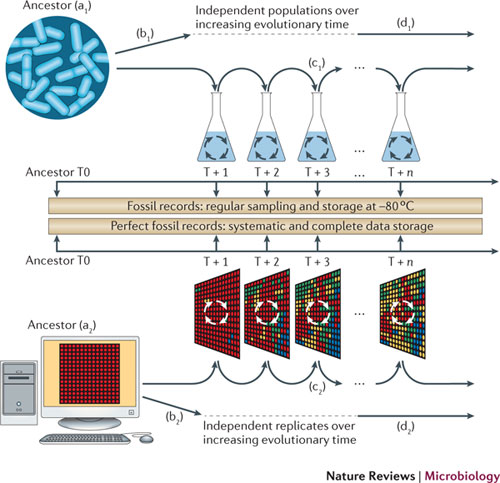
As a consequence, this model can be used to study how evolutionary pressures like the ones for robustness or evolvability can shape genome structure [60] , [57] , [58] , [67] . Indeed, using this model, we have shown that genome compactness is strongly influenced by indirect selective pressures for robustness and evolvability. By genome compactness, we mean several structural features of genome structure, like gene number, amount of non functional DNA, presence or absence of overlapping genes, presence or absence of operons [60] , [57] , [68] . More precisely, we have shown that the genome evolves towards a compact structure if the rate of spontaneous mutations and rearrangements is high. As far as gene number is concerned, this effect was known as an error-threshold effect [51] . However, the effect we observed on the amount of non functional DNA was unexpected. We have shown that it can only be understood if rearrangements are taken into account: by promoting large duplications or deletions, non functional DNA can be mutagenic for the genes it surrounds.
We have extended this framework to include genetic regulation (R-Aevol variant of the model). We are now able to study how these pressures also shape the structure and size of the genetic network in our virtual organisms [44] , [43] , [45] . Using R-Aevol we have been able to show that (i) the model qualitatively reproduces known scaling properties in the gene content of prokaryotic genomes and that (ii) these laws are not due to differences in lifestyles but to differences in the spontaneous rates of mutations and rearrangements [43] . Our approach consists in addressing unsolved questions on Darwinian evolution by designing controlled and repeated evolutionary experiments, either to test the various evolutionary scenarios found in the literature or to propose new ones. Our experience is that “thought experiments” are often misleading: because evolution is a complex process involving long-term and indirect effects (like the indirect selection of robustness and evolvability), it is hard to correctly predict the effect of a factor by mere thinking. The type of models we develop are particularly well suited to provide control experiments or test of null hypotheses for specific evolutionary scenarios. We often find that the scenarios commonly found in the literature may not be necessary, after all, to explain the evolutionary origin of a specific biological feature. No selective cost to genome size was needed to explain the evolution of genome compactness [60] , and no difference in lifestyles and environment was needed to explain the complexity of the gene regulatory network [43] . When we unravel such phenomena in the individual-based simulations, we try to build “simpler” mathematical models (using for instance population genetics-like frameworks) to determine the minimal set of ingredients required to produce the effect. Both approaches are complementary: the individual-based model is a more natural tool to interact with biologists, while the mathematical models contain fewer parameters and fewer ad-hoc hypotheses about the cellular chemistry.
At this time, simulating the evolution of large genomes during hundreds of thousands of generation with the Aevol software can take several weeks or even months. It is worse with Raevol, where we not only simulate mutations and selection at the evolutionary timescale, but also simulate the lifetime of the individuals, allowing them to respond to environmental signals. Previous efforts to parallelize and distribute Aevol had yielded limited results due to the lack of dedicated staff on these problems. Since September 2014, we have been improving the performance of (R-)Aevol. Thanks to the ADT Aevol, one and a half full time engineers work on improving Aevol and especially to parallelize it. Moreover, we are working to formalize the numerical computation problems with (R-)Aevol to use state-of-the-art optimization techniques from the HPC community. It ranges from dense and sparse matrix multiplication and their optimizations (such as Tridiagonal matrix algorithm) to using new generation accelerator (Intel Xeon Phi and NVidia GPU). However, our goal is not to become a HPC nor a numerical computation team but to work with well-established teams in these fields, such as through the Joint Laboratory for Extreme-Scale Computing, but also with Inria teams in these fields (e.g. ROMA, Avalon, CORSE, RUNTIME, MESCAL). By doing so, (R-)Aevol simulations will be faster, allowing us to study more parameters in a shorter time. Furthermore, we will also be able to simulate more realistic population sizes, that currently do not fit into the memory of a single computer.
In 2015 we have improved both the quality and the performance of the code. For example, we are currently testing a new representation of the phenotype allowing us to use vector operation. In collaboration with the Avalon team and with the help of a shared internship (Mehdi Ghesh), we have build a benchmark for ordinary differential equation (ODE). This benchmark is based on a representative sample of the ODEs (formalizing the genetic network) found within the R-Aevol model. Thanks to this benchmark, we can compare different ODE solvers and methods. Furthermore, researchers working on ODE solvers and methods could use it to evaluate the quality of their approach. We are now working with Avalon team on an algorithm that will automatically choose at runtime the best fitting solver and method (from a performance and a quality of results point of view). Through this collaboration, we have also extended the execo experimental engine (Matthieu Imbert, Laurent Pouilloux, Jonathan Rouzaud-Cornabas, Adrien Lèbre, Takahiro Hirofuchi “Using the EXECO toolbox to perform automatic and reproducible cloud experiments” 1st International Workshop on UsiNg and building ClOud Testbeds UNICO, collocated with IEEE CloudCom 2013 2013) to support Aevol and R-Aevol. By doing so, we have now a complete automatic workflow to conduct large scale campaign experiments with thousands of different parameters of our model and use the resources of distributed platform (Grid'5000 in our case).
Since 2014, we are also working on a second model of genome evolution.This new model, developed by the team within the Evoevo european Project, encompasses not only the gene regulation network (as Raevol does) but also the metabolic level [36] . It allows us to have a real notion of resources and thus to have more complex ecological interactions between the individuals. To speed up computations, the genomic level is simplified compared to aevol, as a chromosome is modelled as a sequence of genes and regulatory elements and not as a sequence of nucleotides. Both models are thus complementary.
Little has been achieved concerning the validation of these models, and the relevance of the observed evolutionary tendencies for living organisms. Some comparisons have been made between Avida and experimental evolution [61] , [55] , but the comparison with what happened in a long timescale to life on earth is still missing. It is partly because the reconstruction of ancient genomes from the similarities and differences between extant ones is a difficult computational problem which still misses good solutions for every type of mutations, in particular the ones concerning changes in the genome structure.
There exist good phylogenic models of punctual mutations on sequences [53] , which enable the reconstruction of small parts of ancestral sequences, individual genes for example [62] . But models of whole genome evolution, taking into account large scale events like duplications, insertions, deletions, lateral transfer, rearrangements are just being developped [70] , [49] . Integrative phylogenetic models, considering both nucleotide subsitutions and genome architectures, like Aevol does, are still missing.
Partial models lead to evolutionary hypotheses on the birth and death of genes [50] , on the rearrangements due to duplications [41] , [69] , on the reasons of variation of genome size [56] , [63] . Most of these hypotheses are difficult to test due to the difficulty of in vivo evolutionary experiments.
To this aim, we develop evolutionary models to reconstruct the history of organisms from the comparison of their genome, at every scale, from nucleotide substitutions to genome organisation rearrangements. These models include large-scale duplications as well as loss of DNA material, and lateral gene transfers from distant species. In particular we have developed models of evolution by rearrangements [64] , methods for reconstructing the organization of ancestral genomes [65] , [47] , [66] , or for detecting lateral gene transfer events [40] , [8] . It is complementary with the Aevol development because both the model of artificial evolution and the phylogenetic models we develop emphasize on the architecture of genomes. So we are in a good position to compare artificial and biological data on this point.
We improve the phylogenetic models to reconstruct ancestral genomes, jointly seen as gene contents, orders, organizations, sequences. It requires integrative models of genome evolution, which is desirable not only because they will provide a unifying view on molecular evolution, but also because they will shed light onto the relations between different kinds of mutations, and enable the comparison with artificial experiments from models like Aevol.
Based on this experience, the Beagle team contributes individual-based and mathematical models of genome evolution, in silico experiments as well as historical reconstruction on real genomes, to shed light on the evolutionary origin of the complex properties of cells.