
Keywords
Computer Science and Digital Science
- A2.5. Software engineering
- A3.3.2. Data mining
- A3.4.1. Supervised learning
- A3.4.2. Unsupervised learning
- A6.1.4. Multiscale modeling
- A6.2.4. Statistical methods
- A6.2.8. Computational geometry and meshes
- A8.1. Discrete mathematics, combinatorics
- A8.3. Geometry, Topology
- A8.7. Graph theory
- A9.2. Machine learning
Other Research Topics and Application Domains
- B1.1.1. Structural biology
- B1.1.5. Immunology
- B1.1.7. Bioinformatics
1 Team members, visitors, external collaborators
Research Scientists
- Frédéric Cazals [Team leader, Inria, Senior Researcher, HDR]
- Dorian Mazauric [Inria, Researcher, HDR]
- Edoardo Sarti [Inria, Researcher, from Oct 2021]
Post-Doctoral Fellow
- Vladimir Krajnak [Inria, from Oct 2021]
PhD Student
- Timothee O Donnell [Inria]
Interns and Apprentices
- Louis Goldenberg [Inria, from Mar 2021 until Aug 2021]
- Aarushi Gupta [Inria, from May 2021 until Aug 2021]
- Valentin Madelaine [Inria, until Mar 2021]
- Maximilien Martin [École Normale Supérieure de Lyon, from Oct 2021]
Administrative Assistant
- Florence Barbara [Inria]
External Collaborators
- Charles Robert [CNRS, HDR]
- Konstantin Roeder [Robinson College - Cambridge]
2 Overall objectives
Biomolecules and their function(s).
Computational Structural Biology (CSB) is the scientific domain concerned with the development of algorithms and software to understand and predict the structure and function of biological macromolecules. This research field is inherently multi-disciplinary. On the experimental side, biology and medicine provide the objects studied, while biophysics and bioinformatics supply experimental data, which are of two main kinds. On the one hand, genome sequencing projects give supply protein sequences, and ~200 millions of sequences have been archived in UniProtKB/TrEMBL – which collects the protein sequences yielded by genome sequencing projects. On the other hand, structure determination experiments (notably X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy) give access to geometric models of molecules – atomic coordinates. Alas, only ~150,000 structures have been solved and deposited in the Protein Data Bank (PDB), a number to be compared against the sequences found in UniProtKB/TrEMBL. With one structure for ~1000 sequences, we hardly know anything about biological functions at the atomic/structural level. Complementing experiments, physical chemistry/chemical physics supply the required models (energies, thermodynamics, etc). More specifically, let us recall that proteins with atoms has Cartesian coordinates, and fixing these (up to rigid motions) defines a conformation. As conveyed by the iconic lock-and-key metaphor for interacting molecules, Biology is based on the interactions stable conformations make with each other. Turning these intuitive notions into quantitative ones requires delving into statistical physics, as macroscopic properties are average properties computed over ensembles of conformations. Developing effective algorithms to perform accurate simulations is especially challenging for two main reasons. The first one is the high dimension of conformational spaces – see above, typically several tens of thousands, and the non linearity of the energy functionals used. The second one is the multiscale nature of the phenomena studied: with biologically relevant time scales beyond the millisecond, and atomic vibrations periods of the order of femto-seconds, simulating such phenomena typically requires conformations/frames, a (brute) tour de force rarely achieved 38.
Computational Structural Biology: three main challenges.
The first challenge, sequence-to-structure prediction, aims to infer the possible structure(s) of a protein from its amino acid sequence. While recent progress has been made recently using in particular deep learning techniques 37, the models obtained so far are static and coarse-grained.
The second one is protein function prediction. Given a protein with known structure i.e. 3D coordinates, the goal is to predict the partners of this protein, in terms of stability and specificity. This understanding is fundamental to biology and medicine, as illustrated by the example of the SARS-CoV-2 virus responsible of the Covid19 pandemic. To infect a host, the virus first fuses its envelope with the membrane of a target cell, and then injects its genetic material into that cell. Fusion is achieved by a so-called class I fusion protein, also found in other viruses (influenza, SARS-CoV-1, HIV, etc). The fusion process is a highly dynamic process involving large amplitude conformational changes of the molecules. It is poorly understood, which hinders our ability to design therapeutics to block it.
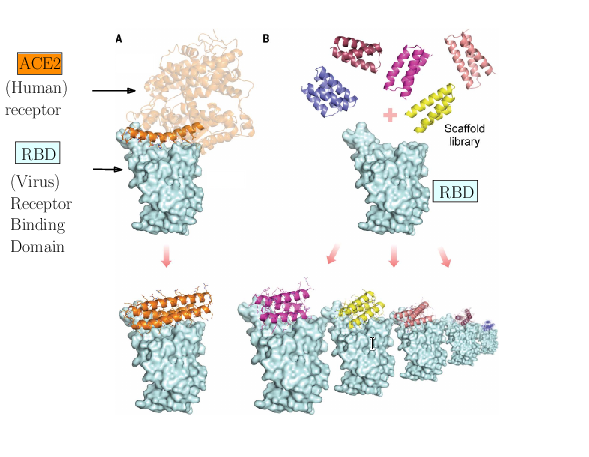
The spike of SARS-CoV-2 is responsible for the infection. Molecules can be engineered to compete with the receptor of the spike, and block infection.
Finally, the third one, large assembly reconstruction, aims at solving (coarse-grain) structures of molecular machines involving tens or even hundreds of subunits. This research vein was promoted about 15 years back by the work on the nuclear pore complex 26. It is often referred to as reconstruction by data integration, as it necessitates to combine coarse-grain models (notably from cryo-electron microscopy (cryo-EM) and native mass spectrometry) with atomic models of subunits obtained from X ray crystallography. Fitting the latter into the former requires exploring the conformation space of subunits, whence the importance of protein dynamics.
As an illustration of these three challenges, consider the problem of designing proteins blocking the entry of SARS-CoV-2 into our cells (Fig. 1). The first challenge is illustrated by the problem of predicting the structure of a blocker protein from its sequence of amino-acids – a tractable problem here since the mini proteins used only comprise of the order of 50 amino-acids (Fig. 1(A), 29). The second challenge is illustrated by the calculation of the binding modes and the binding affinity of the designed proteins for the RBD of SARS-CoV-2 (Fig. 1(B)). Finally, the last challenge is illustrated by the problem of solving structures of the virus with a cell, to understand how many spikes are involved in the fusion mechanism leading to infection. In 29, the promising designs suggested by modeling have been assessed by an array of wet lab experiments (affinity measurements, circular dichroism for thermal stability assessment, structure resolution by cryo-EM). The hyperstable minibinders identified provide starting points for SARS-CoV-2 therapeutics 29. We note in passing that this is truly remarkable work, yet, the designed proteins stem from a template (the bottom helix from ACE2), and are rather small.
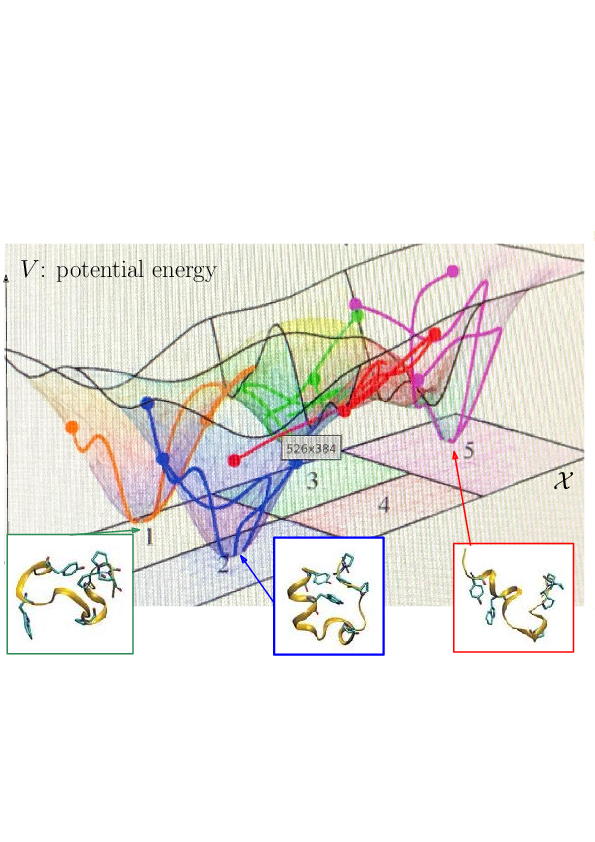
Prosaically, the energy landscape of a molecular system is the set of valleys and passes connecting them, corresponding to stable states and transitions between them.
Protein dynamics: core CS - maths challenges.
To present challenges in structural modeling, let us recall the following ingredients. First, a molecular model with atoms is parameterized over a conformational space of dimension in Cartesian coordinates, or in internal coordinate–upon removing rigid motions, also called degree of freedom (d.o.f.). Second, recall that the potential energy landscape (PEL) is the mapping from to providing a potential energy for each conformation 39, 36. Example potential energies (PE) are CHARMM, AMBER, MARTINI, etc. Such PE belong to the realm of molecular mechanics, and implement atomic or coarse-grain models. They may embark a solvent model, either explicit or implicit. Their definition requires a significant number of parameters (up to ), fitted to reproduce physico-chemical properties of (bio-)molecules 40.
These PE are usually considered good enough to study non covalent interactions – our focus, even tough they do not cover the modification of chemical bonds. In any case, we take such a function for granted 1.
The PEL codes all structural, thermodynamic, and kinetic properties, which can be obtained by averaging properties of conformations over so-called thermodynamic ensembles. The structure of a macromolecular system requires the characterization of active conformations and important intermediates in functional pathways involving significant basins. In assigning occupation probabilities to these conformations by integrating Boltzmann's distribution, one treats thermodynamics. Finally, transitions between the states, modeled, say, by a master equation (a continuous-time Markov process), correspond to kinetics. Classical simulation methods based on molecular dynamics (MD) and Monte Carlo sampling (MC) are developed in the lineage of the seminal work by the 2013 recipients of the Nobel prize in chemistry (Karplus, Levitt, Warshel), which was awarded “for the development of multiscale models for complex chemical systems”. However, except for highly specialized cases where massive calculations have been used 38, neither MD nor MC give access to the aforementioned time scales. In fact, the main limitation of such methods is that they treat structural, thermodynamic and kinetic aspects at once 32. The absence of specific insights on these three complementary pieces of the puzzle makes it impossible to optimize simulation methods, and results in general in the inability to obtain converged simulations on biologically relevant time-scales.
The hardness of structural modeling owes to three intertwined reasons.
First, PELs of biomolecules usually exhibit a number of critical points exponential in the dimension 27; fortunately, they enjoy a multi-scale structure 30. Intuitively, the significant local minima/basins are those which are deep or isolated/wide, two notions which are mathematically qualified by the concepts of persistence and prominence. Mathematically, problems are plagued with the curse of dimensionality and measure concentration phenomena. Second, biomolecular processes are inherently multi-scale, with motions spanning 15 and 4 orders of magnitude in time and amplitude respectively 25. Developing methods able to exploit this multi-scale structure has remained elusive. Third, macroscopic properties of biomolecules i.e. observables, are average properties computed over ensembles of conformations, which calls for a multi-scale statistical treatment both of thermodynamics and kinetics.
Validating models.
A natural and critical question naturally concerns the validation of models proposed in structural bioinformatics. For all three types of questions of interest (structures, thermodynamics, kinetics), there exist experiments to which the models must be confronted – when the experiments can be conducted.
For structures, the models proposed can readily be compared against experimental results stemming from X ray crystallography, NMR, or cryo electron microscopy. For thermodynamics, which we illustrate here with binding affinities, predictions can be compared against measurements provided by calorimetry or surface plasmon resonance. Lastly, kinetic predictions can also be assessed by various experiments such as binding affinity measurements (for the prediction of and ), or fluorescence based methods (for kinetics of folding).
3 Research program
Our research program ambition to develop a comprehensive set of novel concepts and algorithms to study protein dynamics, based on the modular framework of PEL.
3.1 Modeling the dynamics of proteins
Keywords: Molecular conformations, conformational exploration, energy landscapes, thermodynamics, kinetics.
As noticed while discussing Protein dynamics: core CS - maths challenges, the integrated nature of simulation methods such as MD or MC is such that these methods do not in general give access to biologically relevant time scales. The framework of energy landscapes 39, 36 (Fig. 2) is much more modular, yet, large biomolecular systems remain out of reach.
To make a definitive step towards solving the prediction of protein dynamics, we will serialize the discovery and the exploitation of a PEL 4, 13, 3. Ideas and concepts from computational geometry/geometric motion planning, machine learning, probabilistic algorithms, and numerical probability will be used to develop two classes of probabilistic algorithms. The first deals with algorithms to discover/sketch PELs i.e. enumerate all significant (persistent or prominent) local minima and their connections across saddles, a difficult task since the number of all local minima/critical points is generally exponential in the dimension. To this end, we will develop a hierarchical data structure coding PELs as well as multi-scale proposals to explore molecular conformations. (Nb: in Monte Carlo methods, a proposal generates a new conformation from an existing one.) The second focuses on methods to exploit/sample PELs i.e. compute so-called densities of states, from which all thermodynamic quantities are given by standard relations 2835. This is a hard problem akin to high-dimensional numerical integration. To solve this problem, we will develop a learning based strategy for the Wang-Landau algorithm 34–an adaptive Monte Carlo Markov Chain (MCMC) algorithm, as well as a generalization of multi-phase Monte Carlo methods for convex/polytope volume calculations 33, 31, for non convex strata of PELs.
3.2 Algorithmic foundations: geometry, optimization, machine learning
Keywords: Geometry, optimization, machine learning, randomized algorithms, sampling, optimization..
As discussed in the previous Section, the study of PEL and protein dynamics raises difficult algorithmic / mathematical questions. As an illustration, one may consider our recent work on the comparison of high dimensional distribution 6, statistical tests / two-sample tests 7, 10, the comparison of clustering 8, the complexity study of graph inference problems for low-resolution reconstruction of assemblies 9, the analysis of partition (or clustering) stability in large networks, the complexity of the representation of simplicial complexes 2. Making progress on such questions is fundamental to advance the state-of-the art on protein dynamics.
We will continue to work on such questions, motivated by CSB / theoretical biophysics, both in the continuous (geometric) and discrete settings. The developments will be based on a combination of ideas and concepts from computational geometry, machine learning (notably on non linear dimensionality reduction, the reconstruction of cell complexes, and sampling methods), graph algorithms, probabilistic algorithms, optimization, numerical probability, and also biophysics.
3.3 Software: the Structural Bioinformatics Library
Keywords: Scientific software, generic programming, molecular modeling..
While our main ambition is to advance the algorithmic foundations of molecular simulation, a major challenge will be to ensure that the theoretical and algorithmic developments will change the fate of applications, as illustrated by our case studies. To foster such a symbiotic relationship between theory, algorithms and simulation, we will pursue high quality software development and integration within the SBL, and will also take the appropriate measures for the software to be widely adopted.
Software in structural bioinformatics.
Software development for structural bioinformatics is especially challenging, combining advanced geometric, numerical and combinatorial algorithms, with complex biophysical models for PEL and related thermodynamic/kinetic properties. Specific features of the proteins studied must also be accommodated. About 50 years after the development of force fields and simulation methods (see the 2013 Nobel prize in chemistry), the software implementing such methods has a profound impact on molecular science at large. One can indeed cite packages such as CHARMM, AMBER, gromacs, gmin, MODELLER, Rosetta, VMD, PyMol, .... On the other hand, these packages are goal oriented, each tackling a (small set of) specific goal(s). In fact, no real modular software design and integration has taken place. As a result, despite the high quality software packages available, inter-operability between algorithmic building blocks has remained very limited.
The SBL.
Predicting the dynamics of large molecular systems requires the integration of advanced algorithmic building blocks / complex software components. To achieve a sufficient level of integration, we undertook the development of the Structural Bioinformatics Library (SBL, SB) 5, a generic C++/python cross-platform library providing software to solve complex problems in structural bioinformatics. For end-users, the SBL provides ready to use, state-of-the-art applications to model macro-molecules and their complexes at various resolutions, and also to store results in perennial and easy to use data formats (SBL Applications). For developers, the SBL provides a broad C++/python toolbox with modular design (SBL Doc). This hybrid status targeting both end-users and developers stems from an advanced software design involving four software components, namely applications, core algorithms, biophysical models, and modules (SBL Modules). This modular design makes it possible to optimize robustness and the performance of individual components, which can then be assembled within a goal oriented application.
3.4 Applications: modeling interfaces, contacts, and interactions
Keywords: Protein interactions, protein complexes, structure/thermodynamics/kinetics prediction.
Our methods will be validated on various systems for which flexibility operates at various scales. Example such systems are antibody-antigen complexes, (viral) polymerases, (membrane) transporters.
Even very complex biomolecular systems are deterministic in prescribed conditions (temperature, pH, etc), demonstrating that despite their high dimensionality, all d.o.f. are not at play at the same time. This insight suggests three classes of systems of particular interest. The first class consists of systems defined from (essentially) rigid blocks whose relative positions change thanks to conformational changes of linkers; a Newton cradle provides an interesting way to envision such as system. We have recently worked on one such system, a membrane proteins involve in antibiotic resistance (AcrB, see 14. The second class consists of cases where relative positions of subdomains do not significantly change, yet, their intrinsic dynamics are significantly altered. A classical illustration is provided by antibodies, whose binding affinity owes to dynamics localized in six specific loops 11, 12. The third class, consisting of composite cases, will greatly benefit from insights on the first two classes. As an example, we may consider the spikes of the SARS-CoV-2 virus, whose function (performing infection) involves both large amplitude conformational changes and subtle dynamics of the so-called receptor binding domain. We have started to investigate this system, in collaboration with B. Delmas (INRAe) 15.
In ABS, we will investigate systems in these three tiers, in collaboration with expert collaborators, to hopefully open new perspectives in biology and medicine. Along the way, we will also collaborate on selected questions at the interface between CSB and systems biology, as it is now clear that the structural level and the systems level (pathways of interacting molecules) can benefit from one another.
4 Application domains
The main application domain is Computational Structural Biology, as underlined in the Research Program.
5 Highlights of the year
In October 2021, Edoardo Sarti has joined ABS as Chargé de Recherche de Classe Normale. His expertise comprises a diverse set of interests spanning from algorithmic questions about geometrical, functional and evolutionary aspects of biomolecules (latest study: 23), to the collection and analysis of large collections of molecular structural data. From the very start, E. Sarti has started taking part in several research and technical projects of ABS.
6 New software and platforms
See report on the Structural Bioinformatics Library.
6.1 New software
6.1.1 SBL
-
Name:
Structural Bioinformatics Library
-
Keywords:
Structural Biology, Biophysics, Software architecture
-
Functional Description:
The SBL is a generic C++/python cross-platform software library targeting complex problems in structural bioinformatics. Its tenet is based on a modular design offering a rich and versatile framework allowing the development of novel applications requiring well specified complex operations, without compromising robustness and performances.
More specifically, the SBL involves four software components (1-4 thereafter). For end-users, the SBL provides ready to use, state-of-the-art (1) applications to handle molecular models defined by unions of balls, to deal with molecular flexibility, to model macro-molecular assemblies. These applications can also be combined to tackle integrated analysis problems. For developers, the SBL provides a broad C++ toolbox with modular design, involving core (2) algorithms, (3) biophysical models, and (4) modules, the latter being especially suited to develop novel applications. The SBL comes with a thorough documentation consisting of user and reference manuals, and a bugzilla platform to handle community feedback.
-
Release Contributions:
In 2021, two new packages have been released. The first one, Frechet mean on the unit circle ( https://sbl.inria.fr/doc/Frechet_mean_S1-user-manual.html ) provides the first algorithm to compute the exact center of mass of angular data – a key step in computing rotamers for example. The second one, tripeptide loop closure ( https://sbl.inria.fr/doc/Tripeptide_loop_closure-user-manual.html ) is concerned with the reconstruction of all backbone geometries for a tripeptide when the 6 dihedral angles associated with the three Calpha carbons are free to move – all other internal coordinates being fixed. This algorithm is a cornerstone of move sets in internal coordinates.
The SBL has also started a new process of innovation and enhanced distribution. On the one hand, a pre-existing conda package has been revamped, thus extending the range of OS supporting ready-to-use SBL functions to MacOS and Windows. On the other hand, the library has been compiled in a user-ready Singularity container, and can be now used on any Linux system, including environments where the user does not have administrator/root privileges, e.g. clusters and distributed systems for scientific computing.
- URL:
- Publication:
-
Contact:
Frédéric Cazals
7 New results
7.1 Modeling interfaces, contacts, and interactions
Keywords: docking, scoring, interfaces, protein complexes, Voronoi diagrams, arrangements of balls.
7.1.1 Boosting the analysis of protein interfaces with Multiple Interface String Alignments: illustration on the spikes of coronaviruses
Participant: F. Cazals.
In collaboration with S. Bereux and B. Delmas (INRAe, Jouy-en-Josas).
In this work 15, we introduce Multiple Interface String Alignment (MISA), a visualization tool to display coherently various sequence and structure based statistics at protein-protein interfaces (SSE elements, buried surface area, , B factor values, etc). The amino-acids supporting these annotations are obtained from Voronoi interface models. The benefit of MISA is to collate annotated sequences of (homologous) chains found in different biological contexts i.e. bound with different partners or unbound. The aggregated views MISA/SSE, MISA/BSA, MISA/ etc make it trivial to identify commonalities and differences between chains, to infer key interface residues, and to understand where conformational changes occur upon binding. As such, they should prove of key relevance for knowledge based annotations of protein databases such as the Protein Data Bank.
Illustrations are provided on the receptor binding domain (RBD) of coronaviruses, in complex with their cognate partner or (neutralizing) antibodies. MISA computed with a minimal number of structures complement and enrich findings previously reported.
The corresponding package is available from the Structural Bioinformatics Library (SBL
and MISA).
7.1.2 SARS-CoV-2 Through the Lens of Computational Biology: How bioinformatics is playing a key role in the study of the virus and its origins
Participant: F. Cazals.
In collaboration with Samuel Alizon (MIVEGEC - Maladies infectieuses et vecteurs : écologie, génétique, évolution et contrôle), Stéphane Guindon (MAB - Méthodes et Algorithmes pour la Bioinformatique, LIRMM - Laboratoire d'Informatique de Robotique et de Microélectronique de Montpellier), Claire Lemaitre (GenScale - Scalable, Optimized and Parallel Algorithms for Genomics, Inria Rennes – Bretagne Atlantique), Tristan Mary-Huard (INRAE - Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement), Anna Niarakis (Lifeware - Computational systems biology and optimization, Inria Saclay - Ile de France; GenHotel - Laboratoire de recherche européen pour la polyarthrite rhumatoïde), Mikaël Salson (CRIStAL - Centre de Recherche en Informatique, Signal et Automatique de Lille - UMR 9189), Celine Scornavacca (UMR ISEM - Institut des Sciences de l'Evolution de Montpellier), Hélène Touzet (CRIStAL - Centre de Recherche en Informatique, Signal et Automatique de Lille - UMR 9189).
On December 2019, the Chinese Center for Disease Control reported several cases of severe pneumonia that resists usual treatments in the city of Wuhan. This was the beginning of the COVID-19 pandemic which caused more than 80 millions infection cases and 1.7 millions deaths during the year 2020 alone1 . This major outbreak has given rise to global public health responses as well as an international research effort of unprecedented scope and speed. This scientific mobilization has led to remarkable results, which have enabled a great deal of knowledge to be accumulated in just a few months on this novel pathogen: identification of the virus, of its main proteins, analysis of its origin and its functionning. This basic biological knowledge is mandatory to medical advances: design tests, find a vaccine or a cure.
In this document 21, one year after the beginning of the worldwide spread of the disease, we wish to shed particular light on the contribution of bioinformatics in all this work. Bioinformatics is a discipline at crossroads of computer sciences, mathematics and biology that has taken on an inestimable importance in modern biology and medicine. It provides computational models, algorithms and software to the scientific community, that are both operational and effective. The discovery and study of the SARS-Cov-2 coronavirus is an emblematic example. The utilization of bioinformatics methods has been at the heart of essential milestones : from the sequencing of the virus genome and its annotation to the history of its origin, the modelisation of interacting biological entities both at the molecular scale and at the network scale, and the study of the host genetic susceptibility. All these studies, as a whole, have made it possible to elucidate the nature and the functionning of the novel pathogen and have greatly contributed to the fight against COVID-19.
7.1.3 Gene prioritization based on random walks with restarts and absorbing states, to define gene sets regulating drug pharmacodynamics from single-cell analyses
Participant: F. Cazals, D. Mazauric, A. Sales de Queiroz, G. Sales Santa Cruz.
In collaboration with Alain Jean-Marie (Inria Neo) and Jérémie Roux (Inserm and CNRS and UCA).
Prioritizing genes for their role in drug sensitivity, is an important step in understanding drugs mechanisms of action and discovering new molecular targets for co-treatment. In this work 24, we formalize this problem by considering two sets of genes and respectively composing the predictive gene signature of sensitivity to a drug and the genes involved in its mechanism of action, as well as a protein interaction network (PPIN) containing the products of and as nodes. We introduce Genetrank, a method to prioritize the genes in for their likelihood to regulate the genes in .
Genetrank uses asymmetric random walks with restarts, absorbing states, and a suitable renormalization scheme. Using novel so-called saturation indices, we show that the conjunction of absorbing states and renormalization yields an exploration of the PPIN which is much more progressive than that afforded by random walks with restarts only. Using MINT as underlying network, we apply Genetrank to a predictive gene signature of cancer cells sensitivity to tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL), performed in single-cells. Our ranking provides biological insights on drug sensitivity and a gene set considerably enriched in genes regulating TRAIL pharmacodynamics when compared to the most significant differentially expressed genes obtained from a statistical analysis framework alone. We also introduce gene expression radars, a visualization tool to assess all pairwise interactions at a glance.
Genetrank is made available in the Structural Bioinformatics Library (Genetrank). It should prove useful for mining gene sets in conjunction with a signaling pathway, whenever other approaches yield relatively large sets of genes.
7.2 Modeling the dynamics of proteins
Keywords: protein, flexibility, collective coordinate, conformational sampling dimensionality reduction.
7.2.1 Tripeptide loop closure: a detailed study of reconstructions based on Ramachandran distributions
Participant: F. Cazals, T. O'Donnell.
In collaboration with C. Robert (IBPC / CNRS, Paris, France).
Tripeptide loop closure (TLC) is a standard procedure to reconstruct protein backbone conformations, by solving a polynomial system in a single variable yielding up to 16 real solutions.
In this work 17, we first show that multiprecision is required in a TLC solver to guarantee the existence and the accuracy of solutions. We then compare solutions yielded by the TLC solver against tripeptides from the Protein Data Bank. We show that these solutions are geometrically diverse (up to RMSD with respect to the data), and sound in terms of potential energy. Finally, we compare Ramachandran distributions of data and reconstructions for the three amino acids. The distribution of reconstructions in the second angular space stands out, with a rather uniform distribution leaving a central void.
We anticipate that these insights, coupled to our robust implementation in the Structural Bioinformatics Library (TLC), will help understanding the properties of TLC reconstructions, with potential applications to the generation of conformations of flexible loops in particular.
7.3 Algorithmic foundations
Keywords: Computational geometry, computational topology, optimization, data analysis.
7.3.1 Frechet mean and p-mean on the unit circle: characterization, decidability, and algorithm
Participant: F. Cazals, T. O'Donnell.
In collaboration with B. Delmas (INRAe, Jouy-en-Josas).
The center of mass of a point set lying on a manifold generalizes the celebrated Euclidean centroid, and is ubiquitous in statistical analysis in non Euclidean spaces.
In this work 18, we give a complete characterization of the weighted -mean of a finite set of angular values on , based on a decomposition of such that the functional of interest has at most one local minimum per cell. This characterization is used to show that the problem is decidable for rational angular values –a consequence of Lindemann's theorem on the transcendence of , and to develop an effective algorithm parameterized by exact predicates. A robust implementation of this algorithm based on multi-precision interval arithmetic is also presented, and is shown to be effective for large values of and . We use it as building block to implement the k-means and k-means++ clustering algorithms on the flat torus, with applications to clustering protein molecular conformations. These algorithms are available in the Structural Bioinformatics Library (SBL).
Our derivations are of interest in two respects. First, efficient -mean calculations are relevant to develop principal components analysis on the flat torus encoding angular spaces–a particularly important case to describe molecular conformations. Second, our two-stage strategy stresses the interest of combinatorial methods for p-means, also emphasizing the role of numerical issues.
7.3.2 Improved polytope volume calculations based on Hamiltonian Monte Carlo with boundary reflections and sweet arithmetics
Participant: F. Cazals, A. Chevallier.
In collaboration with S. Pion IMS (Univ. Bordeaux / Bordeaux INP / CNRS UMR 5218).
Computing the volume of a high dimensional polytope is a fundamental problem in geometry, also connected to the calculation of densities of states in statistical physics, and a central building block of such algorithms is the method used to sample a target probability distribution.
This paper 16 studies Hamiltonian Monte Carlo (HMC) with reflections on the boundary of a domain, providing an enhanced alternative to Hit-and-run (HAR) to sample a target distribution restricted to the polytope. We make three contributions. First, we provide a convergence bound, paving the way to more precise mixing time analysis. Second, we present a robust implementation based on multi-precision arithmetic, a mandatory ingredient to guarantee exact predicates and robust constructions. We however allow controlled failures to happen, introducing the Sweeten Exact Geometric Computing (SEGC) paradigm. Third, we use our HMC random walk to perform H-polytope volume calculations, using it as an alternative to HAR within the volume algorithm by Cousins and Vempala. The systematic tests conducted up to dimension on the cube, the isotropic and the standard simplex show that HMC significantly outperforms HAR both in terms of accuracy and running time. Additional tests show that calculations may be handled up to dimension . These tests also establish that multiprecision is mandatory to avoid exits from the polytope.
7.3.3 Overlaying a hypergraph with a graph with bounded maximum degree, with application for low-resoluton reconstructions of molecular assemblies
Participant: D. Mazauric.
In collaboration with F. Havet, T. V. H. Nguyen laboratoire I3S (CNRS, Université Côte d'Azur).
We analyze a generalization of the minimum connectivity inference problem (MCI) that models the computation of low-resolution structures of macro-molecular assemblies, based on data obtained by native mass spectrometry. The generalization studied in this work, allows us to consider more refined constraints for the characterization of low resolution models of large assemblies, such as degree constraints (e.g. a protein has a limited number of other proteins in contact).
More precisely, let and be respectively a graph and a hypergraph defined on a same set of vertices, and let be a graph. We say that -overlays a hyperedge of if the subgraph of induced by contains as a spanning subgraph, and that -overlays if it -overlays every hyperedge of . For a fixed graph and a fixed integer , the problem --Overlay consists in deciding whether there exists a graph with maximum degree at most that -overlays a given hypergraph . In 22, we prove that for any graph which is neither complete nor anticomplete, there exists an integer such that --Overlay is -complete for all .
7.3.4 Conflict coloring problems: complexity and application to high resolution biological assembly modeling
Participant: F. Cazals, D. Mazauric.
In collaboration with F. Havet, T. V. H. Nguyen laboratoire I3S (CNRS, Université Côte d'Azur).
Given a graph , a color set for each vertex , a bipartite graph between color sets and for every edge , Conflict Coloring consists in deciding whether exists a conflict coloring, that is a coloring in which is not an edge of the bipartite graph. Conflict Coloring is motivated by computational structural biology problems, high resolution determination of molecular assemblies. The graph represents the subunits and the interaction between them, the colors are the given conformations, and the edges of the bipartite graphs are the incompatible conformations of two subunits.
In this work, we first establish the complexity dichotomies (polynomial vs -complete) for Conflict Coloring and its variants. We provide some experiments in which we build instances of Conflict Coloring associated to Voronoi diagram in the plane, and we then analyse the existences of a solution related to parameters used in our experimental setup.
8 Partnerships and cooperations
Participant: F. Cazals, D. Mazauric.
8.1 International research visitors
8.1.1 Visits of international scientists
Inria International Chair
- David Wales, Cambridge University, is endowed chair within 3IA Côte d'Azur / ABS.
9 Dissemination
9.1 Promoting scientific activities
9.1.1 Scientific events: organisation
Frédéric Cazals was involved in the organization of:
- Symposium Multidisciplinary approaches in cancer research, Organized at Inria Sophia Antipolis Méditerranée. Web: UCANCER2020.
- Winter School Machine Learning Methods to Analyze and Predict Protein Structure, Dynamics and Function, CIRM, Luminy, November 7-12, 2021. Web: AlgoSB 2021.
- Critical evaluation of methods for scoring interfaces of protein complexes, Online Elixir 3D-Bioinfo meeting, organized by Emmanuel Levy (Elixir IL), Frederic Cazals (Elixir FR), Shoshana Wodak (Elixir BE).
9.1.2 Scientific events: selection
Member of the conference program committees
Frédéric Cazals participated to the following program committees:
- Symposium on Solid and Physical Modeling
- Intelligent Systems for Molecular Biology (ISMB) / European Conference on Computational Biology (ECCB)
9.1.3 Invited talks
Frédéric Cazals gave the following invited talks:
- Mining protein flexibility: a new class of move sets; GDR BIM/GT MASIM, November 2021; UCA, 5th Academy 4 Research Webinar - Mental Retardation and Protein Dynamics, October 2021.
9.1.4 Leadership within the scientific community
Frédéric Cazals
- 2010-...: Member of the steering committee of the GDR Bioinformatique Moléculaire, for the Structure and macro-molecular interactions theme.
- 2017-...: Co-chair, with Yann Ponty, of the working group / groupe de travail (GT MASIM - Méthodes Algorithmiques pour les Structures et Interactions Macromoléculaires), within the GDR de BIoinfor- matique Moléculaire (GDR BIM, GDR BIM).
9.1.5 Research administration
Frédéric Cazals
- 2018-...: Member of the bureau du comité des équipes projets.
- 2020-...: Member of the bureau of the EUR Life, Université Côte d’Azur.
Dorian Mazauric
- 2019-...: Member of the comité Plateformes.
9.2 Teaching - Supervision - Juries
9.2.1 Teaching
- 2014–...: Master Data Sciences Program (M2), Department of Applied Mathematics, Ecole Centrale-Supélec; Foundations of Geometric Methods in Data Analysis; F. Cazals and M. Carrière, Inria Sophia / (ABS, DataShape). Web: FGMDA.
- 2021–...: Master Data Sciences & Artificial Intelligence (M1), Université Côte d’Azur; Introduction to machine learning (course practicals); E. Sarti.
- 2021–...: Master Data Sciences & Artificial Intelligence (M2), Université Côte d’Azur; Geometric and topological methods in machine learning; F. Cazals, J-D. Boissonnat and M. Carrière, Inria Sophia / (ABS, DataShape, DataShape); Web: GTML.
- 2021–...: Master Cancérologie et Recherche Translationnelle (M2), Université Côte d’Azur; Binding affinity maturation and protein interaction network analysis: two examples of bioinformatics applications in medicine; F. Cazals.
- 2020–...: Master Sciences du Vivant (M2), parcours Biologie, Informatique, Mathématiques, Université Côte d’Azur; Introduction to statistical physics of biomolecules; F. Cazals.
- 2018–...: Master : Algorithmique et Complexité, 23h30 TD, niveau M1, Polytech Nice Sophia, Université Côte d'Azur, filière Sciences Informatiques, France; Dorian Mazauric.
9.2.2 Supervision
PhD thesis:
- PhD in progress, 3rd year: Timothée O'Donnel, Modeling the influenza polymerase. Université Côte d'Azur. Thesis co-supervised by Frédéric Cazals and Bernard Delmas, INRA Jouy-en-Josas.
- Defended PhD: Thi Viet Ha Nguyen, Graph Algorithms techniques for (low and high) resolution models of large protein assemblies. Université Côte d'Azur. Thesis co-supervised by Frédéric Havet, Laboratoire I3S (CNRS, Université Côte d'Azur).
Interns:
- Aarushi Gupta, intern from IIT Delhi, summer 2021. Modeling protein backbone flexibility using solutions of the tripeptide loop closure.
- Louis Goldenberg, intern from Ecole Polytechnique, summer 2021. Parametric models for compact clusters.
- Sebastián Gallardo Diaz, Universidad Técnica Federico Santa Marı́a, Valparaı́so, Chile. Advisors: Pierre Kornprobst (Inria project-team Biovision), Dorian Mazauric. Algorithms for a new packing problem : Towards Reading Accessible Newspapers.
- Vivian Losciale, Université Côte d'Azur. Advisors: Jérémy Camponovo, Frédéric Havet, Buntheng Ly, Dorian Mazauric, Maxime Sermesant.Jeux-vidéos de médiation : Intelligence artificielle pour l’imagerie médicale.
- Quentin Larose, Université Côte d'Azur. Advisors: Agnès Bessière, Carole Clastres, Jérémy Camponovo, Luc Hogie, Dorian Mazauric, Eric Pascual, Sandrine Selosse, Brigitte Trousse. Portail des ressources Terra Numerica.
9.2.3 Juries
Frédéric Cazals participated to the following committees:
- Luke Dicks, Cambridge University, April 2021. Rapporteur for the PhD thesis K-means landscapes: exploring clustering solution spaces using energy landscape theory. Advisor: David Wales.
- Manon Ruffini, Univ. of Toulouse, March 2021. Rapporteur on the PhD thesis Models and Algorithms for Computational Protein Design. Advisor: Thomas Schiex.
- Dorian Mazauric, Habilitation thesis, Université Côte d'Azur, November 2021. Committee member (president) for the habilitation Algorithmique des graphes pour les réseaux et la biologie structurale computationnelle.
Dorian Mazauric participated to the following committees:
- Thi Viet Ha Nguyen, Université Côte d'Azur, December 2021. Committee member for the PhD thesis Graph Algorithms techniques for (low and high) resolution models of large protein assemblies. Advisors: Frédéric Havet, Dorian Mazauric.
9.3 Popularization
9.3.1 Internal or external Inria responsibilities
Dorian Mazauric:
- 2019–...: Head of Commission Mastic (Médiation et Animation des MAthématiques, des Sciences et Techniques Informatiques et des Communications), Inria Sophia Antipolis - Méditerranée.
- 2019–...: Coordinator of Terra Numerica – vers une Cité du Numérique, an ambitious scientific popularisation project. Its main goal is to create a "Dedicated Digital space" in the south of France, (in the spirit of the "Cité des Sciences" or "Palais de la découverte" in Paris). To do so, Terra Numerica is developing and structuring popularisation activities, supports which are spread in different antennas throughout the territory (e.g. Espace Terra Numerica - Valbonne Sophia Antipolis, MIA, in schools, exhibition extensions...). This large-scale project involves (brings together) all the actors of research, education, industry, associations and collectivities... It is actually composed of more than one hundred people.
- 2018–...: Member of the Conseil d'Administration de l'association les Petits Débrouillards.
- 2017–...: Member of projet de médiation Galéjade : Graphes et ALgorithmes : Ensemble de Jeux À Destination des Ecoliers... (mais pas que).
9.3.2 Articles and contents
Frédéric Cazals:
- Podcast Investiga’Sciences Vive la protéine: interview-discussion of Thomas Schiex and myself by Valérie Ravinet, October 2021. Vive la protéine.
Dorian Mazauric:
- Participation to the development of Terra Numerica Resources.
- Participation to the development of popularization videos games Terra Numerica videos games.
9.3.3 Interventions
Dorian Mazauric - Fête de la Science 2021:
- Village des Sciences de Villeneuve-Loubet Avec Pobot. Samedi 02 octobre 2021 et dimanche 03 octobre 2021. With Thomas Dissaux, Adrien Gausseran, Nicolas Nisse, Eric Pascual, Lucas Picassari-Arrieta, Brigitte Trousse.
- Village des sciences de la vallée de la Vésubie avec Les Apprentis Pas Sages Samedi 02 octobre 2021. Puzzle du nid d’abeilles – Graphes et algorithmes grandeur nature. With Samantha Lanney-Ricci, Magali Martin-Mazauric.
- Interventions au Campus International de Valbonne Lundi 04 octobre 2021. La magie du binaire – Pas besoin de réfléchir, les ordinateurs calculent tellement vite ? Problèmes actuels en algorithmique. With Estelle Zavoli.
- Atelier scientifique à l’Espace d’Art Concret (EAC), Mouans-Sartoux Organisé par l’EAC (Amandine Briand, Sabrina Lah, Martin Merle, Claire Spada, Brigitte Segatori, Roubaud). Du lundi 04 octobre 2021 au vendredi 08 octobre 2021. Des reines sur une oeuvre d’art (Tenth Copper Corner une oeuvre minimaliste de Carl André formée de 55 carreaux) : mathématiques et algorithmique. With Frédéric Havet, Nicolas Nisse, Martine Olivi. En collaboration avec Geoffroy Aubry et Valérie Doya (atelier de Physique).
- Intervention au collège La chênaie de Mouans-Sartoux Mercredi 06 octobre 2021. La magie des graphes et du binaire – Pas besoin de réfléchir, les ordinateurs calculent tellement vite ? Problèmes actuels en algorithmique. With Mylène Raibaudi, Brigitte Trousse.
- Village des Sciences de Mouans-Sartoux Samedi 09 octobre 2021. Sabrina Barnabé, Martine Olivi, Brigitte Trousse, Thierry Viéville.
- Festival des sciences de Nice d’Université Côte d’Azur Samedi 09 octobre 2021 et dimanche 10 octobre 2021. With Alexandre Bonlarron, Foivos Fioravantes, Victor Jung, Hicham Lesfari, Steve Malalel, Magali Martin-Mazauric, Romain Michelucci, Nicolas Nisse, Marie Pelleau, Nina Singlan, Rudan Xiao.
- Interventions au collège de Roquebillière Jeudi 07 octobre 2021. La magie des graphes et du binaire – Jeux combinatoires – Pas besoin de réfléchir, les ordinateurs calculent tellement vite ? Problèmes actuels en algorithmique – Ateliers Jeux Graphes et Algorithmes. With Samantha Lanney-Ricci.
- Conférence à la médiathèque de Biot Vendredi 08 octobre 2021.
- Village des Sciences et de l’Innovation de la CASA à Antibes Juan-les-Pins Avec PoBot, SLV, @b4games. Samedi 16 octobre 2021 et dimanche 17 octobre 2021. With Agnès Bessière, Armel Berceliot, Étienne Chaplain, Thomas Dissaux, Thierry Lespinasse, Stéphane Mansour, Magali Martin-Mazauric, Nicolas Nisse, Eric Pascual, Lucas Picassari-Arrieta, Frédéric Rallo, Sandrine Selosse, Brigitte Trousse.
Dorian Mazauric - Interventions at Maison de l'Intelligence Artificielle:
- Ateliers Terra Numerica avec les étudiants du Master SmartEdTech. Mercredi 14 avril 2021. Journée intensive de formation hybride et animation et co-création d’ateliers. With Saint-Clair Lefevre, Frédéric Havet, Margarida Romero, Thierry Viéville.
Dorian Mazauric - Cordées de la réussite (coordonné par Université Côte d'Azur):
- Deux classes du collège Henri Nans, Aups. Les sciences du numérique à portée de mains ! Découvrir, Explorer, Expérimenter ! Pirates et trésor : des maths et des algorithmes à la programmation Scratch et mBot. With Frédéric Havet, Eric Pascual, Brigitte Trousse.
Dorian Mazauric - Programme Chiche:
- Intervention au lycée Apollinaire, Nice Jeudi 14 octobre 2021.
- Intervention au lycée Estienne d’Orves, Nice Jeudi 21 octobre 2021.
- Intervention au CIV, Valbonne Sophia Antipolis Jeudi 02 décembre 2021.
Dorian Mazauric - Formations:
- Formation d’enseignants co-organisée par la DANE et Terra Numerica avec les ateliers Terra Numerica à la Maison de l’intelligence Artificielle. Mardi 9 mars 2021, mardi 23 mars 2021, mardi 6 avril 2021, mardi 20 avril 2021, mardi 25 mai 2021. Machine d’apprentissage par renforcement pour gagner aux jeux, Initiation à la reconnaissance d’images avec des drones, ateliers d’informatique débranchée. With Jérémy Camponovo, Frédéric Havet, Eric Pascual, Brigitte Trousse.
- Formation de personnels de médiathèques de la CASA. Vendredi 25 juin 2021, jeudi 23 septembre 2021. Formation sur les fondements de l’informatique : Transmission de pensée – La magie du binaire.
- Présentation et formation au Fab’Ecole 06 de la DRANE, collège Bertone d’Antibes. Vendredi 26 novembre 2021. Présentation et formation sur des ateliers Terra Numerica. With Brigitte Trousse.
Dorian Mazauric - In schools:
- Collège Bechet d’Antibes Juan-les-Pins Lundi 8 mars 2021. Dans le cadre du projet pédagogique Ethique des données et de l’information (1/3). Introductions aux algorithmes. With Sylvain Etienne, Frédéric Giroire, Géraldine Rouard, Brigitte Trousse.
- Centre International de Valbonne Sophia Antipolis Lundi 15 mars 2021. Dans le cadre de séances autour de l’Intelligence Artificielle avec une classe de terminale du CIV organisées par Les Petits Débrouillards. Intelligence Artificielle et reconnaissance d’images. With Marie Barbieux, Marine Beaudet, Soledad Tolosa.
- Collège Bechet d’Antibes Juan-les-Pins Vendredi 26 mars 2021 et 9 avril 2021. Dans le cadre du projet pédagogique Ethique des données et de l’information (2/3). Modélisation d’un réseau social et de contenus, et algorithmes de recommandation. With Sylvain Etienne, Frédéric Giroire, Géraldine Rouard, Brigitte Trousse.
- Collège Bechet d’Antibes Juan-les-Pins Lundi 7 juin 2021. Dans le cadre du projet pédagogique Ethique des données et de l’information (3/3). Conférence Protection des données et métier de Déléguée à la Protection des Données d’Inria (Anne Combe). With Anne Combe, Sylvain Etienne, Frédéric Giroire, Géraldine Rouard, Brigitte Trousse.
- Roquefort-les-Pins Dans le cadre des activités du centre aéré de la commune. Lundi 26 juillet 2021 et mardi 27 juillet 2021. Trois demi-journées : ateliers d’informatique débranchée (pour les 3 à 6 ans), ateliers pour découvrir les algorithmes de recommandation dans les réseaux sociaux (pour les adolescents) et tours de magie pour découvrir comment l’ordinateur compte (pour les 6 à 10 ans). With Frédéric Havet.
- Lycée Internationale de Valbonne Jeudi 02 décembre 2021. Ateliers algorithmiques grandeur nature. With Bérengère Abric, Perrine Le Dûs.
Dorian Mazauric - Internships:
- Treize stagiaires de troisième au centre Inria d’Université Côte d’Azur Du lundi 13 décembre au vendredi 17 décembre 2021.
10 Scientific production
10.1 Major publications
- 1 articleDistributed Link Scheduling in Wireless Networks.Discrete Mathematics, Algorithms and Applications1252020, 1-38
- 2 articleOn the complexity of the representation of simplicial complexes by trees.Theoretical Computer Science617February 2016, 17
- 3 articleEnergy landscapes and persistent minima.The Journal of Chemical Physics14452016, 4URL: https://www.repository.cam.ac.uk/handle/1810/253412
- 4 articleConformational Ensembles and Sampled Energy Landscapes: Analysis and Comparison.J. of Computational Chemistry36162015, 1213--1231URL: https://hal.science/hal-01076317
- 5 reportThe Structural Bioinformatics Library: modeling in biomolecular science and beyond.RR-8957InriaOctober 2016
- 6 inproceedingsBeyond Two-sample-tests: Localizing Data Discrepancies in High-dimensional Spaces.IEEE/ACM International Conference on Data Science and Advanced AnalyticsIEEE/ACM International Conference on Data Science and Advanced AnalyticsIEEE/ACM International Conference on Data Science and Advanced AnalyticsParis, FranceMarch 2015, 29
- 7 inproceedingsLow-Complexity Nonparametric Bayesian Online Prediction with Universal Guarantees.NeurIPS 2019 - Thirty-third Conference on Neural Information Processing SystemsVancouver, CanadaDecember 2019
- 8 articleComparing Two Clusterings Using Matchings between Clusters of Clusters.ACM Journal of Experimental Algorithmics241December 2019, 1-41
- 9 articleComplexity dichotomies for the Minimum F -Overlay problem.Journal of Discrete Algorithms52-53September 2018, 133-142
- 10 articleA Sequential Non-Parametric Multivariate Two-Sample Test.IEEE Transactions on Information Theory645May 2018, 3361-3370
- 11 reportHigh Resolution Crystal Structures Leverage Protein Binding Affinity Predictions.RR-8733InriaMarch 2015
- 12 articleNovel Structural Parameters of Ig–Ag Complexes Yield a Quantitative Description of Interaction Specificity and Binding Affinity.Frontiers in Immunology8February 2017, 34
- 13 articleHybridizing rapidly growing random trees and basin hopping yields an improved exploration of energy landscapes.J. Comp. Chem.3782016, 739--752URL: https://hal.inria.fr/hal-01191028
- 14 articleStudying dynamics without explicit dynamics: A structure‐based study of the export mechanism by AcrB.Proteins - Structure, Function and BioinformaticsSeptember 2020
10.2 Publications of the year
International journals
International peer-reviewed conferences
Doctoral dissertations and habilitation theses
Reports & preprints
10.3 Cited publications
- 25 articleMolecular dynamics: survey of methods for simulating the activity of proteins.Chemical reviews10652006, 1589--1615
- 26 articleThe molecular architecture of the nuclear pore complex.Nature45071702007, 695--701
- 27 articleDynamics on statistical samples of potential energy surfaces.The Journal of chemical physics11151999, 2060--2070
- 28 bookThermodynamics and an Introduction to Thermostatistics.Wiley1985
- 29 articleDe novo design of picomolar SARS-CoV-2 miniprotein inhibitors.Science37065152020, 426--431
- 30 articleEnergy landscapes and persistent minima.The Journal of Chemical Physics14452016, 4URL: https://www.repository.cam.ac.uk/handle/1810/253412
- 31 articleA practical volume algorithm.Mathematical Programming Computation822016, 133--160
- 32 bookUnderstanding molecular simulation.Academic Press2002
-
33
articleRandom walks and an
) volume algorithm for convex bodies.Random Structures & Algorithms1111997, 1--50 - 34 bookA guide to Monte Carlo simulations in statistical physics.Cambridge university press2014
- 35 bookFree energy computations: A mathematical perspective.World Scientific2010
- 36 articlePrediction, determination and validation of phase diagrams via the global study of energy landscapes.Int. J. of Materials Research10022009, 135
- 37 articleImproved protein structure prediction using potentials from deep learning.Nature2020, 1--5
- 38 articleAtomic-level characterization of the structural dynamics of proteins..Science33060022010, 341--346URL: http://dx.doi.org/10.1126/science.1187409
- 39 bookEnergy Landscapes.Cambridge University Press2003
- 40 articleBuilding force fields: an automatic, systematic, and reproducible approach.The journal of physical chemistry letters5112014, 1885--1891