
2023Activity reportProject-TeamDRACULA
RNSR: 201020961V- Research center Inria Lyon Centre
- In partnership with:Université Claude Bernard (Lyon 1), CNRS
- Team name: Multi-scale modelling of cell dynamics : application to hematopoiesis
- In collaboration with:Institut Camille Jordan
- Domain:Digital Health, Biology and Earth
- Theme:Modeling and Control for Life Sciences
Keywords
Computer Science and Digital Science
- A6.1. Methods in mathematical modeling
- A6.1.1. Continuous Modeling (PDE, ODE)
- A6.1.2. Stochastic Modeling
- A6.1.3. Discrete Modeling (multi-agent, people centered)
- A6.1.4. Multiscale modeling
- A6.2.1. Numerical analysis of PDE and ODE
- A6.2.3. Probabilistic methods
- A6.2.4. Statistical methods
- A6.3.1. Inverse problems
Other Research Topics and Application Domains
- B1.1.2. Molecular and cellular biology
- B1.1.3. Developmental biology
- B1.1.4. Genetics and genomics
- B1.1.5. Immunology
- B1.1.6. Evolutionnary biology
- B1.1.7. Bioinformatics
- B1.1.8. Mathematical biology
- B1.1.10. Systems and synthetic biology
- B2.2.1. Cardiovascular and respiratory diseases
- B2.2.3. Cancer
- B2.2.5. Immune system diseases
- B2.2.6. Neurodegenerative diseases
1 Team members, visitors, external collaborators
Research Scientists
- Mostafa Adimy [Team leader, INRIA, Senior Researcher, HDR]
- Samuel Bernard [CNRS, Researcher, HDR]
- Clément Erignoux [INRIA, ISFP, from Dec 2023]
- Olivier Gandrillon [CNRS, Senior Researcher, HDR]
- Thomas Lepoutre [INRIA, Researcher, HDR]
Faculty Members
- Thibault Espinasse [UNIV LYON I, Associate Professor]
- Laurent Pujo-Menjouet [UNIV LYON I, Associate Professor, HDR]
- Leon Tiné [UNIV LYON I, Associate Professor]
Post-Doctoral Fellows
- Mathieu Calero [UNIV LYON I, Post-Doctoral Fellow, from Oct 2023]
- Thi Nhu Thao Nguyen [INRIA, Post-Doctoral Fellow]
- Nicolas Torres [UNIV LYON I, Post-Doctoral Fellow, until Mar 2023]
PhD Students
- Charlotte Camus [INRIA]
- Kyriaki Dariva [UNIV Jean Monnet, ATER, until Aug 2023]
- Maxime Estavoyer [INRIA]
- Clemence Fournie [CNRS, from Oct 2023]
- Grégoire Ranson [UNIV YORK]
Interns and Apprentices
- Manon Charmasson [INRIA, Intern, from Apr 2023 until Jul 2023]
Administrative Assistant
- Claire Sauer [INRIA]
2 Overall objectives
2.1 Presentation
Dracula is a joint research team between INRIA, Université Claude Bernard Lyon 1 (UCBL) and CNRS (Institut Camille-Jordan (ICJ, UMR 5208) and Laboratoire de Biologie et Modélisation de la Cellule (LBMC, UMR 5239)).
The Dracula project is devoted to multi-scale modeling in biology and medicine, and more specifically to the development of tools and methods to describe multi-scale processes in biology and medicine. Applications include normal and pathological hematopoiesis (for example leukemia), immune response, and other biological processes, like: tissue renewal, morphogenesis, atherosclerosis, prion disease, hormonal regulation of food intake, and so on. Multi-scale modeling implies simultaneous modeling of several levels of descriptions of biological processes: intra-cellular networks (molecular level), cell behavior (cellular level), dynamics of cell populations (organ or tissue) with the control by other organs (organism) (see Figure 1). Such modeling represents one of the major challenges in modern science due to its importance and because of the complexity of biological phenomena and of the presence of very different interconnected scales.
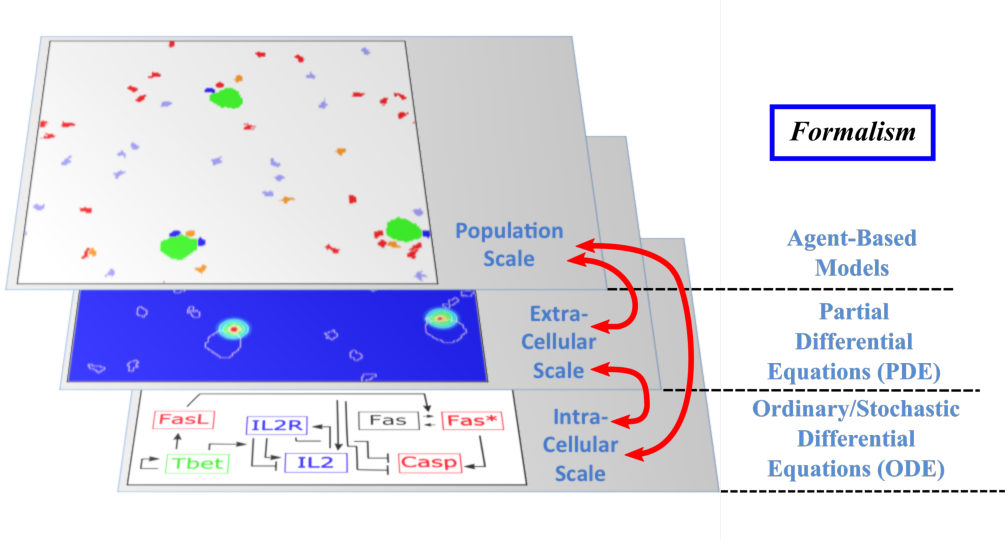
Scheme of multi-scale models of cell dynamics
Although multi-scale modeling holds a great potential for biology and medicine, and despite the fact that a variety of techniques exists to deal with such problems, the complexity of the systems poses new challenges and needs the development of new tools. Moreover, different biological questions usually require different types of multi-scale modeling. The expected results of these studies are numerous. On one hand, they will shed new light on the understanding of specific biological and medical questions (for instance, what is the behavior of hematopoietic stem cells under pathological conditions? Or how to efficiently stimulate an immune response in order to design new vaccines?). On the other hand, the modeling methods developed here for specific processes are relevant to study other complex biological systems. We pay a special attention on developing methods that are not restricted to one or two applications.
An important part of our researches is performed in close collaboration with biologists and physicians in order to stay in contact with the biological and medical goals. The presence, within the project, of a biologist (Olivier Gandrillon) who has acquired over the years the know-how required for interacting with mathematicians is probably one of the main assets of the project. He participates actively in many tasks of our program, stimulates interactions between members of the project and biologists, and everyone benefits from his expertise in molecular and cell biology.
2.2 Keywords
Multi-scale modeling; Hybrid modeling; Mathematical Biology; Computational Biology; Immune response modeling; Normal and pathological hematopoiesis; Multi-scale cancer modeling; Regulatory networks; Reaction-diffusion equation; Structured partial differential equations; Delay differential equations; Agent-based modeling; Dynamical systems.
2.3 Research axis 1: Mathematical modeling for cell population dynamics
Executive summary
Stem cells are essential for development and keep the maintenance of many tissues homeostasis. They are characterized by their ability to self-renew as well as to produce differentiated cells. They vary enormously, for each organ, in their proliferation capacity, their potency to produce different cell lineage and their response to various environmental cues. How a cell will react to a given external signal does not depend only on its current state but also on its environment. Understanding the effect of cell-to-cell heterogeneity and the spatial organization of cell populations is therefore necessary to help keeping the normal function of an organ.
We develop mathematical tools and methods to study cell population dynamics and other biological processes: stability of steady sates, existence of bifurcations, kinetic properties, spatial organization, in finely detailed cell populations. The main tools we use are hybrid discrete-continuous models, reaction-diffusion equations, structured models (in which the population is endowed with relevant structures or traits), delay differential systems, agent-based models. Our team has acquired an international expertise in the fields of analysis of reaction-diffusion and structured equations, particularly integro-differential and delay differential equations.
The mathematical methods we develop are not restricted to hematopoietic system (Research axis 2), and immune response (Research axis 3), rather we apply them in many other biological phenomena, for example: tissue renewal, morphogenesis, prion disease, atherosclerosis, hormonal regulation of food intake, cancer, and others.
Project-team positioning
The focus of this objective is the development, analysis and application of hybrid discrete-continuous, reaction-diffusion and structured partial differential models. The structured equations allow a fine description of a population as some structures (age, maturity, intracellular content) change with time. In many cases, structured equations can be partially integrated to yield integro-differential equations (ordinary or partial differential equations involving non-local integral terms), time-delay differential or time-delay partial differential, or coupled differential-difference models. Analysis of integro-differential and time-delay systems deals with existence of solutions and their stability. Applications are found in the study of normal and pathological hematopoietic system (Research axis 2), immune response (Research axis 3), morphogenesis, prion disease, cancer development and treatment, and generally in tissue renewal problems. Models based on structured equations are especially useful to take into account the effect of finite time cells take to divide, die or become mature. Reaction-diffusion equations are used in order to describe spatial distribution of cell populations. It is a well developed area of research in our team which includes qualitative properties of travelling waves for reaction-diffusion systems with or without delay, and complex nonlinear dynamics.
Our team has developed a solid expertise in mathematical analysis of reaction-diffusion with or without delay and structured equations (in particular, delay differential equations) and one of the most prolific. Other major groups are the teams of Benoit Perthame (Pierre et Marie CURIE University and Mamba, Paris), Emmanuel Grenier (Ecole normale supérieure de Lyon and NUMED), Odo Diekmann (Utrecht University, The Netherlands), Avner Friedman (The Ohio State University, USA), Jianhong Wu (York University, Canada), Glenn Webb (Vanderbilt University, Nashville, USA), Philip K. Maini (University of Oxford, England), Mark Chaplain (University of St Andrews, Scotland), Nicola Bellomo (University of Turin, Italy). Most of the members of all these groups and of our team belong to the same mathematical community working on partial differential equations and dynamical systems with applications to biology and medicine.
Collaborations
- University of Toronto, Canada; Mathematical analysis and applications of reaction-diffusion equations (more than 30 joint papers).
- Institute of Problems of Mechanical Engineering, St.Petersburg, Russia; Dynamics of cell renewal (more than 10 joint papers).
- Department of Cell and Molecular Biology and Department of Forensic Medicine, Stockholm, Sweden; Dynamics of cell generation and turnover (3 joint papers).
- Universities of Tlemcen (Algeria) and Marrakech (Morocco); Delay differential equations (7 joint papers)
2.4 Research axis 2: Multi-scale modeling of hematopoiesis and leukemia
Executive summary
Hematopoiesis is a complex process that begins with hematopoietic stem cells (HSCs) and results in formation of mature cells: red blood cells, white cells and platelets. Blood cells are produced in the bone marrow, from where mature cells are released into the blood stream. Hematopoiesis is based on a balance between cell proliferation (including self-renewal), differentiation and apoptosis. The choice between these three possibilities is determined by intra-cellular regulatory networks and by numerous control mechanisms in the bone marrow or carried out by other organs. Intra-cellular regulatory networks are complex biochemical reactions involving proteins, enzymes and signalling molecules. The deregulation of hematopoiesis can result in numerous blood diseases including leukemia (a cancer of blood cells). One important type of leukemia is Chronic Myeloid Leukemia (CML). The strong tyrosine kinase activity of the BCR-ABL protein is the basis for the main cell effects that are observed in CML: significant proliferation, anti-apoptotic effect, disruption of stroma adhesion properties. This explains the presence in CML blood of a very important number of cells belonging to the myeloid lineage, at all stages of maturation.
Multi-scale modeling in hematopoiesis holds a great potential. A variety of techniques exists to deal with this problem. However, the complexity of the system poses new difficulties and leads to the development of new tools. The expected results of this study are numerous. On one hand, it will shed new light on the different physiological mechanisms that converge toward the continuous regeneration of blood cells, for example: the understanding of deregulation of erythropoiesis (the process of red blood cell production) under drug treatments (this can lead to lack of red blood cells (anemia), or a surplus of red blood cells), the dynamic of leukemic cells under the action of drugs and the control of their resistance to these treatments.
Project team positioning
Multi-scale modeling of hematopoiesis is one of the key points of the project that has started in the early stage of the Dracula team. Investigated by all the team members, it took many years of close discussion with biologists to get the best understanding of the key role played by the most important molecules, hormones, kinase cascade, cell communication up to the latest knowledge. One of the important questions here is to identify particular biological mechanisms (intracellular regulation, control mechanisms) and to integrate them in the different models. Our main work consisted in the development of a hybrid (continuous/discrete) model for red blood cell progenitor proliferation, survival/death, differentiation, and migration. Cells are modeled as discrete objects, and the extracellular medium is described by continuous equations for extracellular concentrations. This is to our knowledge the most complete model for erythropoiesis to date, and the only one using a multi-scale formalism. Other models published by our group and others for hematopoiesis are population-based models, mostly population structured equations (transport partial differential equations or delay differential equations). The interest in modeling hematopoiesis dates back to the 70's and two groups have been responsible for most of development in the past 40 years: Markus Loeffer's team in Leipzig, Germany (Wichmann et al. 1976, in Mathematical Models in Medicine) and Michael Mackey's team at McGill University, Montreal, Canada (Mackey 1978, Blood). Our model differs from population based models in that the regulation is directly modeled at the molecular level (See Figure 1) rather than acting on rates at the population level. Thus we can take into account non-predictable effects of interactions between different molecular pathways and between cells that would otherwise be lost in the global population rates.
Regarding modeling leukemia, we concentrated on Chronic Myeloid Leukemia (CML) and its treatment. We considered models based on ordinary differential equations for the action of the main proteins involved in CML (as BCR-ABL protein), and of transport equations (with or without delay, physiologically structured or not) to represent healthy and leukemic cell populations, take into account many interactions between proteins (especially BCR-ABL), cells (anti-apoptotic effect, etc.). The development of models for CML allowed us to interact with Franck Nicolini in Lyon (Centre Hospitalier de Lyon) and Doron Levy (Maryland University). Different schools developed models for CML and its treatment. The three leading groups are the ones of Franziska Michor (Harvard School of public health), Ingo Roeder (Institute for Medical Informatics and Biometry, Dresden) and Michael Mackey (McGill University).
Collaborations
Members of the team have worked for several years in collaboration with biologists (François Morlé, University Lyon 1) and hematologists (Charles Dumontet, Lyon and Mark Koury, Nashville) on the Modelling of normal and pathological hematopoiesis .
The work on modeling Leukemia is based on two major collaborations: firstly, an ongoing (since 2011) mathematical collaboration with the University of Maryland through the program Associate Teams Inria project, “Modelling Leukemia”. Secondly, an ongoing (since 2012) collaboration with a clinician from Hospices Civils de Lyon (Dr. F.E. Nicolini). In this framework, we shall have soon access to the data of the clinical trial PETALs ( patients).
2.5 Research axis 3: Multi-scale modeling of the immune response
Executive summary
Vaccination represents a worldwide health, social and economical challenge as it has allowed the eradication or the strong containment of several devastating diseases over the past century. However to date, most of the effective vaccines rely on the generation of neutralizing antibody responses and such vaccines have proven largely unsuccessful in the prevention against some pathogens, such as HIV or malaria. In such cases, vaccines geared towards the generation of CD8 T cell immunity may provide a better protection. The generation of memory CD8 T cells following antigenic immunization is a long process (lasting up to month in murine preclinical models), therefore strongly slowing the process of vaccine monitoring in preclinical studies. Thus, the dynamical modeling of the CD8 T cell immune response both at the cellular and molecular levels should provide an important tool to better understand the dynamics of the response and to speed-up the process and reduce costs of vaccine development.
However, currently published cellular models of the immune response are either over-simplified, not predicting important parameters of this response, or too complicated for most of their parameters to be accessible for experimental measurements, thus impeding their biological validation. Dynamical models of the CD8 T cell response at the molecular level are very scarce and there is no multi-scale model of the immune response giving insights into both the regulation at the molecular scale and the consequences on cell population dynamics.
The objective of this research axis is therefore to develop a predictive multi-scale model of the CD8 T cell response, by confronting the model at different stages to in vivo-acquired experimental data, in order to be able to investigate the influence of early molecular events on cell population dynamics few days or weeks later.
Project-team positioning
We are aiming at building and analyzing a multi-scale model of the CD8 T cell immune response, from the molecular to the cellular and potentially organismal scale. This consists in describing the dynamics at each scale with relevant formalisms as well as the careful description of the couplings between scales.
Only few research groups are actually working on the CD8 T cell immune response around the world, and none of them deals with multi-scale modeling of this response. A network developed around Alan Perelson's work in theoretical immunology in the last decades, at Los Alamos National Laboratory, and involves mainly people in various US universities or institutes. In Europe, Rob De Boer's group of theoretical immunology in Utrecht, Netherlands, is the historical leader in the CD8 T cell dynamics modeling. We considered the models developed in these groups when we started our project, and we contributed to improve them by using nonlinearities accounting for cell population interactions to regulate the response. Also, our initial focus was on the generation of memory cells associated with vaccine development so we modeled CD8 T cell responses against influenza and vaccinia viruses, whereas other groups usually consider LCMV in its chronic form.
Ron Germain's group at the NIH, and Grégoire Altan-Bonnet in subsequent works, focused on the molecular regulation of the CD4 and CD8 T cell immune responses. In particular, they built the Simmune software, which allows the modeling and simulation of molecular interactions. This software is not really devoted to multi-scale modeling yet it provides an interesting tool to describe molecular interactions. Since our aim is to couple molecular and cellular scales at the tissue level, and we do not want to consider large networks but rather small-simplified informative interaction networks, we are confident that our approach is complementary of these works.
Within Inria project-teams, NUMED develops multi-scale approaches for biological problems, and MAMBA and MONC mention models of cancer progression and treatment including immune responses. In the first case the methodology is similar, and collaborations between NUMED and DRACULA already exist (both teams are located in Lyon), but applications differ. In the second case, MAMBA and MONC are mainly focused on cancer modeling and up to now are motivated by including an action of the immune system in the fight against cancer, which is very different from what we are developing. However, both modeling approaches are complementary and could lead to interactions, in particular in the light of recent advances in medical research pointing towards an important role - and high expectations - of the immune reaction in fighting cancers. Finally, SISTM also focuses on the modeling of the immune response, mainly against HIV, but the motivation is very similar to ours: the objective is to provide tools and methods in order to efficiently develop vaccines. They consider the CD4 T cell response instead of the CD8 T cell response, and biostatistics to achieve their goals instead of multi-scale models, yet even though there is no interaction between SISTM and DRACULA at this moment our methods and objectives are close enough to foreshadow future collaborations.
Collaborations
On this topic our main collaborators are members of Jacqueline Marvel's team in Lyon in the CIRI (Centre International de Recherche en Infectiologie INSERM U1111): Dr. Jacqueline Marvel, head of the team, Dr. Christophe Arpin (CR CNRS), and other technicians and engineers of the team. They are all immunologists, specialists of the CD8 T cell response and of the generation of memory CD8 T cells.
We also interact with private companies: AltraBio, that provides tools for data analysis, and CosmoTech, that develops a modeling and simulating platform that should allow transferring our model on an easy-to-use platform devoted to commercial uses.
2.6 Evolution of research direction during the last evaluation
Reminder of the objectives given for the last evaluation
The aim of this project is the development of modern tools for multi-scale modeling in biological phenomena. During the period 2014-2017, the objectives we had fixed were to develop modern tools for multi-scale modeling of biological phenomena, as detailed hereafter:
- Multi-scale modeling of erythropoiesis, the process of red blood cell production, in order to describe normal, stress, and pathological erythropoiesis, using mathematical and computational models. This led to:
- Multi-scale modeling of the CD8 T cell immune response, in order to develop a predictive model of the CD8 T cell response, by confronting the model at different stages to in vivo-acquired experimental data;
- Population dynamics modeling, with the aim to develop general mathematical tools to study them. The main tools we were using were structured equations, in which the cell population is endowed with relevant structures, or traits. We identified limitations in using these formalisms, this is why we started developing multi-scale approaches;
- Modeling of Chronic Myeloid Leukemia (CML) treatment, using ordinary differential equations models. Our team had already developed a first model of mutant leukemic cells being resistant to chemotherapy. A next step would be to identify the parameters using experimental data;
- Multi-scale modeling carried out on the basis of hybrid discrete-continuous models, where dissipative particle dynamics (DPD) are used in order to describe individual cells and relatively small cell populations, partial differential equations (PDE) are used to describe concentrations of bio-chemical substances in the extracellular matrix, and ordinary differential equations for intracellular regulatory networks (Figure 1). An emphasis would be made on developing codes that are both flexible and powerful enough to implement variants of the model, perform simulations, produce desired outputs, and provide tools for analysis; to do so:
- We planned to contribute to a recent project named chronos, whose code (written in C++) represents heterogeneous populations of individual cells evolving in time and interacting physically and biochemically, and the objective is to make the code flexible enough to implement different formalisms within the same model, so that different components of the model can be represented in the most appropriate way;
- Partial differential equations (PDE) analysis, with a focus on reaction-diffusion equations, transport equations (hyperbolic PDEs) in which the structure can be age, maturity, protein concentration, etc., with particular cases where transport equations are reduced to delay differential equations (DDE).
Comments on these objectives over the evaluation period
We have had strong contributions to objectives 1, 2, 3, 4, and consequently to objective 5, as well as to objective 7, as mentioned in previous sections. These contributions represented the core of the team's research activity over the evaluation period, as stressed by our publications. It is however noticeable that multi-scale modeling of the immune response and of pathological hematopoiesis (leukemia) has come to represent a proportionally more important part of our activity.
Objective 6 has been pursued, the project chronos evolved to a better defined project SiMuScale that is currently being developed and aims at structuring the team's activity and providing a simulation platform that could be adapted to various biological questions necessitating multi-scale modeling.
Objectives for the next four years
The main objectives for the next four years are to continue to improve the 3 previous points: 1) Mathematical and computational modeling for cell population dynamics; 2) Multi-scale modeling of hematopoiesis and leukemia; 3) Multi-scale modeling of the immune response. In addition, we will pursue our effort to develop a simulation platform for multi-scale models (SiMuScale) and we intend to develop the use of mixed effect models and other statistical approaches to deal with the challenges offered by modern biology, in particular the generation of single cell data.
3 Research program
3.1 Mixed-effect models and statistical approaches
Most of biological and medical data that our team has to deal with consist in time series of experimental measurements (cell counts, gene expression level, etc.). The intrinsic variability of any biological system complicates its confrontation to models. The trivial use of means, eliminating the data variance, is but a second-best solution. Furthermore, the amount of data that can be experimentally generated often limits the use of classical mathematical approaches because model’s identifiability or parameter identifiability cannot be obtained. In order to overcome this issue and to efficiently take advantage of existing and available data, we plan to use mixed effect models for various applications (for instance: leukemia treatment modeling, immune response modeling). Such models were initially developed to account for individual behaviors within a population by characterizing distributions of parameter values instead of a unique parameter value. We plan to use those approaches both within that frame (for example, taking into account longitudinal studies on different patients, or different mice) but also to extend its validity in a different context: we will consider different ex vivo experiments as being “different individuals”: this will allow us to make the most of the experience-to-experience variations.
Such approaches need expertise in statistics to be correctly implemented, and we will rely on the presence of Céline Vial in the team to do so. Céline Vial is an expert in applied statistics and her experience already motivated the use of better statistical methods in various research themes. The increasing use of single cell technologies in biology make such approaches necessary and it is going to be critical for the project to acquire such skills.
3.2 Development of a simulation platform
We have put some effort in developing the SiMuScale platform, a software coded in dedicated to exploring multiscale population models, since 2014. In order to answer the challenges of multi-scale modeling it is necessary to possess an all-purpose, fast and flexible modeling tool, and SiMuScale is the choice we made. Since it is based on a core containing the simulator, and on plug-ins that contain the biological specifications of each cell, this software will make it easier for members of the team – and potentially other modelers – to focus on the model and to capitalize on existing models, which all share the same framework and are compatible with each other. Within the next four years, SiMuScale should be widely accessible and daily used in the team for multi-scale modeling. It will be developed into a real-case context, the modeling of the hematopoietic stem cell niche, in collaboration with clinicians (Eric Solary, INSERM) and physicists (Bertrand Laforge, UPMC).
3.3 Mathematical and computational modeling
Multi-scale modeling of hematopoiesis is one of the key points of the project that has started in the early stage of the Dracula team. Investigated by the team members, it took many years of close discussion with biologists to get the best understanding of the key role played by the most important molecules, hormones, kinase cascade, cell communication up to the latest knowledge. An approach that we used is based on hybrid discrete-continuous models, where cells are considered as individual objects, intracellular regulatory networks are described with ordinary differential equations, extracellular concentrations with diffusion or diffusion-convection equations (see Figure 1). These modeling tools require the expertise of all team members to get the most qualitative satisfactory model. The obtained models will be applied particularly to describe normal and pathological hematopoiesis as well as immune response.
3.4 From hybrid dynamics to continuum mechanics
Hybrid discrete-continuous methods are well adapted to describe biological cells. However, they are not appropriate for the qualitative investigation of the corresponding phenomena. Therefore, hybrid model approach should be combined with continuous models. If we consider cell populations as a continuous medium, then cell concentrations can be described by reaction-diffusion systems of equations with convective terms. The diffusion terms correspond to a random cell motion and the reaction terms to cell proliferation, differentiation and death. We will continue our studies of stability, nonlinear dynamics and pattern formation. Theoretical investigations of reaction-diffusion models will be accompanied by numerical simulations and will be applied to study cell population dynamic.
3.5 Structured partial differential equations
Hyperbolic problems are also of importance when describing cell population dynamics. They are structured transport partial differential equations, in which the structure is a characteristic of the considered population, for instance age, size, maturity, etc. In the scope of multi-scale modeling, protein concentrations as structure variables can precisely indicate the nature of cellular events cells undergo (differentiation, apoptosis), by allowing a representation of cell populations in a multi-dimensional space. Several questions are still open in the study of this problem, yet we will continue our analysis of these equations by focusing in particular on the asymptotic behavior of the system (stability, oscillations) and numerical simulations.
3.6 Delay differential equations
The use of age structure in PDE often leads to a reduction (by integration over the age variable) to delay differential equations. Delay differential equations are particularly useful for situations where the processes are controlled through feedback loops acting after a certain time. For example, in the evolution of cell populations the transmission of control signals can be related to some processes as division, differentiation, maturation, apoptosis, etc. Delay differential equations offer good tools to study the behavior of the systems. Our main investigation will be the effect of perturbations of the parameters, as cell cycle duration, apoptosis, differentiation, self-renewal, etc., on the behavior of the system, in relation for instance with some pathological situations. The mathematical analysis of delay differential equations is often complicated and needs the development of new criteria to be performed.
3.7 Multi-scale modeling of the immune response
The main objective of this part is to develop models that make it possible to investigate the dynamics of the adaptive CD8 T cell immune response, and in particular to focus on the consequences of early molecular events on the cellular dynamics few days or weeks later: this would help developing predictive tools of the immune response in order to facilitate vaccine development and reduce costs. This work requires a close and intensive collaboration with immunologist partners.
We recently published a model of the CD8 T cell immune response characterizing differentiation stages, identified by biomarkers, able to predict the quantity of memory cells from early measurements, 21. In parallel, we improved our multiscale model of the CD8 T cell immune response, by implementing a full differentiation scheme, from naïve to memory cells, based on a limited set of genes and transcription factors.
Our first task will be to infer an appropriate gene regulatory network (GRN) using single cell data analysis (generate transcriptomics data of the CD8 T cell response to diverse pathogens), the previous biomarkers we identified and associated to differentiation stages, as well as piecewise-deterministic Markov processes (Ulysse Herbach's PhD thesis, ongoing).
Our second task will be to update our multiscale model by first implementing the new differentiation scheme we identified 21, and second by embedding CD8 T cells with the GRN obtained in our first task (see above). This will lead to a multi-scale model incorporating description of the CD8 T cell immune response both at the molecular and the cellular levels (Simon Girel's PhD thesis, ongoing).
In order to further develop our multiscale model, we will consider an agent-based approach for the description of the cellular dynamics. Yet, such models, coupled to continuous models describing GRN dynamics, are computationally expensive, so we will focus on alternative strategies, in particular on descriptions of the cellular dynamics through both continuous and discrete models, efficiently coupled. Using discrete models for low cell numbers and continuous (partial differential equations) models for large cell numbers, with appropriate coupling strategies, can lead to faster numerical simulations, and consequently can allow performing intense parameter estimation procedures that are necessary to validate models by confronting them to experimental data, both at the molecular and cellular scales.
The final objective will be to capture CD8 T cell responses in different immunization contexts (different pathogens, tumor) and to predict cellular outcomes from molecular events.
3.8 Dynamical network inference from single-cell data
Up to now, all of our multiscale models have incorporated a dynamical molecular network that was build “by hand” after a thorough review of the literature. It would be highly valuable to infer it directly from gene expression data. However, this remains very challenging from a methodological point of view. We started exploring an original solution for such inference by using the information contained within gene expression distributions. Such distributions can be acquired through novel techniques where gene expression levels are quantified at the single cell level. We propose to view the inference problem as a fitting procedure for a mechanistic gene network model that is inherently stochastic and takes not only protein, but also mRNA levels into account. This approach led to very encouraging results 22 and we will actively pursue in that direction, especially in the light of the foreseeable explosion of single cell data.
3.9 Leukemia modeling
Imatinib and other tyrosine kinase inhibitors (TKIs) have marked a revolution in the treatment of Chronic Myelogenous Leukemia (CML). Yet, most patients are not cured, and must take their treatment for life. Deeper mechanistic understanding could improve TKI combination therapies to better control the residual leukemic cell population. In a collaboration with the Hospital Lyon Sud and the University of Maryland, we have developed mathematical models that integrate CML and an autologous immune response (18, 19 and 20). These studies have lent theoretical support to the idea that the immune system plays a rôle in maintaining remission over long periods. Our mathematical model predicts that upon treatment discontinuation, the immune system can control the disease and prevent a relapse. There is however a possibility for relapse via a sneak-though mechanism 18. Research in the next four years will focus in the Phase III PETALS trial. In the PETALS trial, the second generation TKI Nilotinib is combined with Peg-IFN, an interferon that is thought to enhance the immune response. We plan to: 1) Adapt the model to take into account the early dynamics (first three months). 2) Use a mixed-effect approach to analyse the effect of the combination, and find population and individual parameters related to treatment efficacy and immune system response. 3) Optimise long-term treatment strategies to reduce or cease treatment and make personalised predictions based on mixed-effect parameters, to minimise the long-term probability of relapse.
4 Application domains
See the previous sections.
5 New results
Participants: [ All team members.
5.1 Single-cell transcriptional uncertainty landscape of cell differentiation
Background: Single-cell studies have demonstrated the presence of significant cell-to-cell heterogeneity in gene expression. Whether such heterogeneity is only a bystander or has a functional role in the cell differentiation process is still hotly debated. Methods: In this study, 11, we quantified and followed single-cell transcriptional uncertainty – a measure of gene transcriptional stochasticity in single cells – in 10 cell differentiation systems of varying cell lineage progressions, from single to multi-branching trajectories, using the stochastic two-state gene transcription model. Results: By visualizing the transcriptional uncertainty as a landscape over a two-dimensional representation of the single-cell gene expression data, we observed universal features in the cell differentiation trajectories that include: (i) a peak in single-cell uncertainty during transition states, and in systems with bifurcating differentiation trajectories, each branching point represents a state of high transcriptional uncertainty; (ii) a positive correlation of transcriptional uncertainty with transcriptional burst size and frequency; (iii) an increase in RNA velocity preceding the increase in the cell transcriptional uncertainty. Conclusions: Our findings suggest a possible universal mechanism during the cell differentiation process, in which stem cells engage stochastic exploratory dynamics of gene expression at the start of the cell differentiation by increasing gene transcriptional bursts, and disengage such dynamics once cells have decided on a particular terminal cell identity. Notably, the peak of single-cell transcriptional uncertainty signifies the decision-making point in the cell differentiation process.
5.2 One model fits all: Combining inference and simulation of gene regulatory networks
The rise of single-cell data highlights the need for a nondeterministic view of gene expression, while offering new opportunities regarding gene regulatory network inference. We recently introduced two strategies that specifically exploit time-course data, where single-cell profiling is performed after a stimulus: HARISSA, a mechanistic network model with a highly efficient simulation procedure, and CARDAMOM, a scalable inference method seen as model calibration. In 15, we combine the two approaches and show that the same model driven by transcriptional bursting can be used simultaneously as an inference tool, to reconstruct biologically relevant networks, and as a simulation tool, to generate realistic transcriptional profiles emerging from gene interactions. We verify that CARDAMOM quantitatively reconstructs causal links when the data is simulated from HARISSA, and demonstrate its performance on experimental data collected on in vitro differentiating mouse embryonic stem cells. Overall, this integrated strategy largely overcomes the limitations of disconnected inference and simulation.
5.3 A multigroup approach to delayed prion production
We generalize in 5 the model proposed in [Adimy, Babin, Pujo-Menjouet, SIAM Journal on Applied Dynamical Systems (2022)] for prion infection to a network of neurons. We do so by applying a so-called multigroup approach to the system of Delay Differential Equations (DDEs) proposed in the aforementioned paper. We derive the classical threshold quantity , i.e. the basic reproduction number, exploiting the fact that the DDEs of our model qualitatively behave like Ordinary Differential Equations (ODEs) when evaluated at the Disease Free Equilibrium. We prove analytically that the disease naturally goes extinct when , whereas it persists when . We conclude with some selected numerical simulations of the system, to illustrate our analytical results.
5.4 Implication of lipid turnover for the control of energy balance
The ongoing obesity epidemic is a consequence of a progressive energy imbalance. The energy-balance model (EBM) posits that obesity results from an excess in food intake and circulating fuels. A reversal in causality has been proposed recently in the form of the carbohydrate–insulin model (CIM), according to which fat storage drives energy imbalance. Under the CIM, dietary carbohydrates shift energy use in favour of storage in adipose tissue. The dynamics of lipid storage and mobilization could, therefore, be sensitive to changes in carbohydrate intake and represent a measurable component of the CIM. To characterize potential changes in lipid dynamics induced by carbohydrates, mathematical models were used, 6. Here, we propose a coherent mathematical implementation of the CIM-energy deposition model (CIM-EDM), which includes lipid turnover dynamics. Using lipid turnover data previously obtained by radiocarbon dating, we build two cohorts of virtual patients and simulate lipid dynamics during ageing and weight loss. We identify clinically testable lipid dynamic parameters that discriminate between the CIM-EDM and an energy in, energy out implementation of the EBM (EBM-IOM). Using a clinically relevant two-month virtual trial, we additionally identify scenarios and propose mechanisms whereby individuals may respond differently to low-carbohydrate diets. This article is part of a discussion meeting issue ‘Causes of obesity: theories, conjectures and evidence (Part II)’.
5.5 Toward an Early Diagnosis for Alzheimer’s Disease Based on the Perinuclear Localization of the ATM Protein.
Alzheimer's disease (AD) is the most common neurodegenerative dementia, for which the molecular origins, genetic predisposition and therapeutic approach are still debated. In the 1980s, cells from AD patients were reported to be sensitive to ionizing radiation. In order to examine the molecular basis of this radiosensitivity, the ATM-dependent DNA double-strand breaks (DSB) signaling and repair were investigated, in 7, by applying an approach based on the radiation-induced ataxia telangiectasia-mutated (ATM) protein nucleoshuttling (RIANS) model. Early after irradiation, all ten AD fibroblast cell lines tested showed impaired DSB recognition and delayed RIANS. AD fibroblasts specifically showed spontaneous perinuclear localization of phosphorylated ATM (pATM) forms. To our knowledge, such observation has never been reported before, and by considering the role of the ATM kinase in the stress response, it may introduce a novel interpretation of accelerated aging. Our data and a mathematical approach through a brand-new model suggest that, in response to a progressive and cumulative stress, cytoplasmic ATM monomers phosphorylate the APOE protein (pAPOE) close to the nuclear membrane and aggregate around the nucleus, preventing their entry in the nucleus and thus the recognition and repair of spontaneous DSB, which contributes to the aging process. Our findings suggest that pATM and/or pAPOE may serve as biomarkers for an early reliable diagnosis of AD on any fibroblast sample.
5.6 Adaptation of a quantitative trait to a changing environment: new analytical insights on the asexual and infinitesimal sexual models
Predicting the adaptation of populations to a changing environment is crucial to assess the impact of human activities on biodiversity. Many theoretical studies have tackled this issue by modeling the evolution of quantitative traits subject to stabilizing selection around an optimal phenotype, whose value is shifted continuously through time. In this context, the population fate results from the equilibrium distribution of the trait, relative to the moving optimum. Such a distribution may vary with the shape of selection, the system of reproduction, the number of loci, the mutation kernel or their interactions. In 12, we develop a methodology that provides quantitative measures of population maladaptation and potential of survival directly from the entire profile of the phenotypic distribution, without any a priori on its shape. We investigate two different systems of reproduction (asexual and infinitesimal sexual models of inheritance), with various forms of selection. In particular, we recover that fitness functions such that selection weakens away from the optimum lead to evolutionary tipping points, with an abrupt collapse of the population when the speed of environmental change is too high. Our unified framework allows deciphering the mechanisms that lead to this phenomenon. More generally, it allows discussing similarities and discrepancies between the two systems of reproduction, which are ultimately explained by different constraints on the evolution of the phenotypic variance. We demonstrate that the mean fitness in the population crucially depends on the shape of the selection function in the infinitesimal sexual model, in contrast with the asexual model. In the asexual model, we also investigate the effect of the mutation kernel and we show that kernels with higher kurtosis tend to reduce maladaptation and improve fitness, especially in fast changing environments.
5.7 Ergodicity of the Fisher infinitesimal model with quadratic selection
We study in 8 the convergence towards a unique equilibrium distribution of the solutions to a time-discrete model with non-overlapping generations arising in quantitative genetics. The model describes the dynamics of a phenotypic distribution with respect to a multi-dimensional trait, which is shaped by selection and Fisher's infinitesimal model of sexual reproduction. We extend some previous works devoted to the time-continuous analogues, that followed a perturbative approach in the regime of weak selection, by exploiting the contractivity of the infinitesimal model operator in the Wasserstein metric. Here, we tackle the case of quadratic selection by a global approach. We establish uniqueness of the equilibrium distribution and exponential convergence of the renormalized profile. Our technique relies on an accurate control of the propagation of information across the large binary trees of ancestors (the pedigree chart), and reveals an ergodicity property, meaning that the shape of the initial datum is quickly forgotten across generations. We combine this information with appropriate estimates for the emergence of Gaussian tails and propagation of quadratic and exponential moments to derive quantitative convergence rates. Our result can be interpreted as a generalization of the Krein-Rutman theorem in a genuinely non-linear, and non-monotone setting.
5.8 Influence of the age structure on the stability in a tumor-immune model for chronic myeloid leukemia
We propose and analyze in 9 a system of partial differential equations (PDEs) for chronic myeloid leukemia (CML), generalizing the ordinary differential equations' system (ODE) proposed in [2,3]. This model describes the proliferation and differentiation of leukemic cells in the bone marrow and the interactions of leukemic and immune cells. We consider that the differentiation of cells can be described by a continuous variable which we use to structure our system. The model is based on a non-monotonic immune response. At low levels, immune response increases with the tumor load whereas for high levels, tumor is suppressing the effect of immune system (immunosuppression). We analyze the stability of the steady states of the model and compare it to the case of [2] where maturity was described as a discrete state. In particular, a steady state describing remission induced by a control due to the immune system is shown to be unstable in certain situations for the PDE model , whereas in [2] it was systematically stable.
5.9 Asymmetric attractive zero-range processes with particle destruction at the origin
We investigate in 10 the macroscopic behavior of asymmetric attractive zero-range processes on where particles are destroyed at the origin at a rate of order , where and is the scaling parameter. We prove that the hydrodynamic limit of this particle system is described by the unique entropy solution of a hyperbolic conservation law, supplemented by a boundary condition depending on the range of . Namely, if , then the boundary condition prescribes the particle current through the origin, whereas if , the destruction of particles at the origin has no macroscopic effect on the system and no boundary condition is imposed at the hydrodynamic limit.
5.10 Influenza transmissibility among patients and health-care professionals in a geriatric short-stay unit using individual contact data
Detailed information are lacking on influenza transmissibility in hospital although clusters are regularly reported. In this pilot study, 13, our goal was to estimate the transmission rate of H3N2 2012-influenza, among patients and health care professionals in a short-term Acute Care for the Elderly Unit by using a stochastic approach and a simple susceptible-exposed-infectious-removed model. Transmission parameters were derived from documented individual contact data collected by Radio Frequency IDentification technology at the epidemic peak. From our model, nurses appeared to transmit infection to a patient more frequently with a transmission rate of 1.04 per day on average compared to 0.38 from medical doctors. This transmission rate was 0.34 between nurses. These results, even obtained in this specific context, might give a relevant insight of the influenza dynamics in hospitals and will help to improve and to target control measures for preventing nosocomial transmission of influenza. The investigation of nosocomial transmission of SARS-COV-2 might gain from similar approaches.
5.11 Exact hydrodynamics and onset of phase separation for an active exclusion process
We consider in 14 a lattice model of active matter with exclusion and derive its hydrodynamic description exactly. The hydrodynamic limit leads to an integrodifferential equation for the density of particles with a given orientation. Volume exclusion results in nonlinear mobility dependent on spatial density. Such models of active matter can support motility-induced phase separation, which occurs despite the absence of attractive interactions. We study the onset of phase separation with linear stability analysis and numerical simulations.
5.12 Simuscale: A Modular Framework for Multiscale Single-Cell Modelling
Simuscale is a multiscale, individual-based modelling platform for performing numerical simulations of heterogeneous populations of individual cells evolving in time and interacting physically and biochemically, 16. Models are described at two levels: cellular level and population level. The cellular level describes the dynamics of single cells, as defined by the modeller. Cells have an internal state that includes default properties such as cell size and position, and may also include any other cell-specific state, such as gene or protein expression. The population level describes the mechanical constraints and biochemical interactions between cells. Cells evolve in bounded 3D domain, and can divide or die. Simuscale implements the physical simulator that manages the simulations at the population level. It delegates the details of cellular dynamics to each cell. This makes Simuscale modular, as it can accommodate any number of cell models with the same simulation, including models with different modelling formalisms. Biochemical interactions occur between cells that are in contact with each other, through intercellular signals. Intercellular signals can be known to all or to a subset of the cells only. Simuscale expects an input file describing the initial cell population and numerical options, it runs a simulation over a specified time interval, updating the cell population at given time steps, and generates an output file containing the state of each cell at each time step, and the tree of cell divisions and deaths.
5.13 Global Asymptotic Stability of a Hybrid Differential–Difference System Describing SIR and SIS Epidemic Models with a Protection Phase and a Nonlinear Force of Infection
We study in 2 the local and global asymptotic stability of the two steady-states, disease-free and endemic, of hybrid differential-difference SIR and SIS epidemic models with a nonlinear force of infection and a temporary phase of protection against the disease, e.g. by vaccination or medication. The initial model is an age-structured system that is reduced using the method of characteristic lines to a hybrid system, coupled between differential equations and a time continuous difference equation. We first prove that the solutions of the original system can be obtained from the reduced one. We then focus on the reduced system to obtain new results on the asymptotic stability of the two steady-states. We determine the local asymptotic stability of the two steady-states by studying the associated characteristic equation. We then discuss their global asymptotic stability in various situations (SIR, SIS, mass action, nonlinear force of infection), by constructing appropriate Lyapunov functions.
5.14 Global asymptotic stability for a distributed delay differential-difference system of a Kermack-McKendrick SIR model
We investigate in 3 a system of distributed delay differential-difference equations describing an epidemic model of susceptible, infected, recovered and temporary protected population dynamics. A nonlocal term (distributed delay) appears in this model to describe the temporary protection period of the susceptible individuals. We investigate mathematical properties of the model. We obtain the global asymptotic stability of the two steady states: disease-free and endemic. We construct appropriate Lyapunov functionals where the basic reproduction number appears as a threshold for the global asymptotic behavior of the solution between disease extinction and persistence.
5.15 Global stability of a SEIR discrete delay differential‐difference system with protection phase
We consider in 4 an epidemiological model with the four classical compartments of susceptible, exposed, infected, and recovered population. We add a new compartment that is supposed to describe, for a limited time, individuals that are protected from the epidemic through vaccination or medication, for instance. We model the protection phase by an age‐structured partial differential equation. The age is the time since an individual entered the protection phase. The model is then reduced by integration on the characteristics to a differential‐difference system with delay. The discrete delay represents the limited duration of the protection phase. After establishing the basic properties of the model, we show that the disease‐free equilibrium (DFE) is globally asymptotically stable when the basic reproduction number is less than one and is unstable when this number is greater than one. Furthermore, we show that even if there is no mortality during the protection phase and the basic reproduction number is greater than one, the endemic equilibrium is globally asymptotically stable. The proofs of the global asymptotic stability of both equilibria are based on carefully constructed Lyapunov functions. To complete this study on the global dynamics, we discuss some results on weak and strong uniform persistence of the disease. Finally, numerical simulations are performed to illustrate and complete our main results.
6 Bilateral contracts and grants with industry
6.1 Bilateral Grants with Industry
Participants: [ M. Bouvier, [ T. Espinasse, [ O. Gandrillon, [ T. Lepoutre.
- CIFRE PhD for Matteo Bouvier, with the Vidium company. Objective: Identification and control of GRNs. A first step will consist in establishing the proof of concept of a Design Of Experiment method to refine the inference of GRNs. A second step will consist in developing a method for controlling the dynamic stochastic systems that underlie GRNs behavior.
7 Partnerships and cooperations
7.1 International initiatives
7.1.1 Inria associate team not involved in an IIL or an international program
Participants: Mostafa Adimy, Charlotte Dugourd-Camus.
Associated team MoCoVec "Modelling and Biological Control of Vector-Borne Diseases: the case of Malaria and Dengue", 2022-2025. Partner Institution(s): MAMBA Inria, Paris (P.A. Bliman [Coordinator]) - FAPESP São Paulo, Brazil. Focusing on dengue and malaria, two diseases transmitted by vector mosquito and which cause high morbidity and mortality around the world, this project aims to model disease transmission, its spread and control, in a context of climatic and environmental change. For this, the main drives of disease transmission will be addressed to understand which factors modulate the spatio-temporal patterns observed, especially in Brazil.
7.1.2 STIC/MATH/CLIMAT AmSud projects
Participants: Mostafa Adimy.
MATH AmSud project TOMENADE "Topological methods and non autonomous dynamics for delay differential equations", 2022-2024. Partner Institution(s): Laboratoire des Signaux et Systèmes (L2S), CNRS, Centrale Supelec - Argentina (P. Amster [Coordinator]) - Chile - Brazil. This project addresses open problems about non autonomous systems of delay differential equations modeling some phenomena from life sciences, namely, a meta-populations version of the Nicholson equations and models of competition in a stirred chemostat. Nevertheless, the ideas and methods could be certainly extended in several ways, and we also expect to make progress in topics as the (non autonomous) topological linearization problem and the possibility of converse results for persistence.
7.1.3 Participation in other International Programs
Participants: Laurent Pujo-Menjouet, Léon Tine.
ECOS-Sud France-Chilie, C20E03 “Coarsening dynamics: numerical and theoretical analysis of the Lifchitz-Slyozov equation with nucleation and applications to biology”, 2021-2023. Partner Institution(s): Inria ANACONDA - Institut Denis Poisson, University Orléans - Universidad del Bio-Bio, Concepcion, Chilie (E. Hingant, [Coordinator]).
The general objectives of this project are i) To push-forward theoretical studies on well-posedness of solutions of the Lifshitz- Slyozov model and variants, and understand long-time behavior and scaling properties of those solutions, ii) To explore various numerical strategies to accurately simulate solutionsoft heLifshitz-Slyozov model, iii) To apply our developed methodology on biology applications, in particular within interdisciplinary work on protein aggregation in neurodegenerative diseases, and on adipocyte formation dynamics.
Participants: Laurent Pujo-Menjouet, Samuel Bernard, Léon Tine.
PHC-Maghreb “Mathematical modeling and control of the spread of infectious diseases in the Mediterranean area”, 2022-2024. Partner Institution(s): Inria-Dracula, ICJ (Université Lyon 1), Mohammed V University of Rabat, Ecole Nationale d'Ingénieurs de Tunis, Université Tlemcen This research project aims to contribute to the development of some decision-making tools in the context of major global public health challenges: namely the modelling, monitoring and control of the spread of infectious diseases in the socio-economic current context. To do this, it is a question of contributing to: 1) improving mathematical representations of the human immune system, 2) construction of compartmental epidemiological models in which susceptible individuals are divided into several classes according to their immunity (vaccinated or not, age, infection with or without provisional immunity, infection with or without recurrence, etc.).
Participants: Laurent Pujo-Menjouet.
Mecan'os “Si tu ne peux pas l’avoir, (re)construis le !” 2021-2023 Partner Institution(s): Institut Camille Jordan - CUNY, New-York - University of Tlemcen, Algeria. The aim of this project is to describe the gene regulatory network involving osteoblasts and its interactions with key proteins in the mineralization of the collagen matrix. A feedback loop from the matrix stiffness induces whole control of the proteins. The question is how. The application is to be able to produce bio-materials showing any required stiffness properties for bone substitutes for instance.
Participants: Laurent Pujo-Menjouet.
micrOS-macrOS “Modélisation multi-échelle de l'os”, 2021-2023 Participants (Dracula): L. Pujo-Menjouet [Coordinator]. Partner Institution(s): Institut Camille Jordan - CUNY, New-York - University of Tlemcen, Algeria.
The objective of this project is to understand the impact of the gene regulatory network (at the molecular scale) on the bio-mechanic properties of the bone (bone scale). This new multi-scale approach will allow to finely understand the qualitative and quantitative role of osteoblasts and osteoclasts on the normal or pathological bone formation.
7.2 National initiatives
7.2.1 ANR
ANR Prio Diff
-
Title:
Impact of replication and structural diversification of prions on their cerebral dissemination
-
Partner Institution(s):
- Inrae Jouy-en-Josas, France
- Inrae Toulouse, France
- CEA, Fontenay aux Roses, France
- Institut Camille Jordan, France
-
Date/Duration:
July 2021-June 2025
-
Additionnal info/keywords:
Prions are lethal proteinaceous pathogens with major public-health risks due to their zoonotic and iatrogenic potential. They are composed of aggregated, misfolded conformers of the host-encoded prion protein that progressively deposit in the brain by a self-perpetuating reaction. The underlying molecular mechanisms of replication and tissue dissemination remain mostly elusive. Our objective is to model these processes entirely based on recent advances that prion aggregates are conformationally heterogeneous and dynamic rather than uniform and static. To achieve this, we will map in prion-infected brain the structural diversification-to-bioactivity/neurotoxicity landscape of prion assemblies in a spatiotemporal manner and mathematically build a multiscale model of diversification and lesion spreading. The goal is to generate an open access model capable of predicting the disease progression and identify key elementary process for therapeutics intervention and early diagnostics. / Keywords: interactions hôtes pathogènes, infectiologie, modélisation prion, tissue diffusion, neurotropism.
ANR SingleStatOmics
-
Title:
Statistics and Machine Learning for Single Cell Genomics
-
Partner Institution(s):
- University Montpellier
- University Dauphine
- Ecole Central de Nantes
- Google Brain / Mines ParisTech
- AgroParisTech / INRAe
- University Grenoble
- University Lyon 1 (F. Picard [coordinator])
- Ecole Centrale de Lyon.
-
Date/Duration:
2019-2023
-
Additionnal info/keywords:
The goal is to develop new methodologies to investigate cell identity and the dynamics of cell differentiation, by integrating single cell expression and epigenomic data.
ANR Memoires
-
Title:
Multiscale Modeling of CD8 T cell immune responses
-
Partner Institution(s):
- ICL-CIRI (J. Marvel [Coordinator])
- Ecole Centrale de Lyon
- AltraBio.
-
Date/Duration:
2019-2023
-
Additionnal info/keywords:
The main goal will is to identify, at the single cell level, signatures associated with functionally relevant memory cell subsets, to characterize dynamical molecular networks that are predictive of their generation, and to build a multi-scale computational model of this process.
ANR PLUME
-
Title:
Molecular and morphogenetic control of feather pattern formation
-
Partner Institution(s):
- Centre interdisciplinaire de recherche en biologie (M. Manceau [Coordinator])
- Institut de Biologie du Développement de Marseille
- Brandeis University.
-
Date/Duration:
2021-2025
-
Additionnal info/keywords:
The objectives are to identify the hierarchy of pattern forming mechanisms establishing initial patterning spaces, characterise material properties and mechanical stresses controlling primordia self-organisation, and uncover how how cell and mechanical dynamics control the timely wave of primordia production. This work will shed light on the mechanisms governing pattern variation and fidelity in nature.
7.2.2 Other projects
Digitbio
-
Title:
MP DIGIT-BIO, AMI 2021
-
Partner Institution(s):
- Inrae Jouy-en-Josas, France
- Inria, Dracula team, France
-
Date/Duration:
September 2021-August 2023
-
Additionnal info/keywords:
Ce projet a pour but de permettre d’explorer pour la première fois dans le champ des maladies neurodégénératives comment la dynamique des assemblages de PrPSc régit la dissémination, la ségrégation et l’accumulation de certaines sous-populations d’assemblages de PrPSc et conduit à l’apparition d’une structure souche-spécifique ainsi que de possibles fronts de propagation / Interactions hôtes pathogènes, infectiologie, modélisation prion, tissue diffusion, neurotropism.
8 Scientific production
8.1 Major publications
8.2 Publications of the year
International journals
Reports & preprints
8.3 Cited publications
- 18 articleStability Analysis of a Model of Interaction Between the Immune System and Cancer Cells in Chronic Myelogenous Leukemia.Bulletin of Mathematical Biology2017, 1--27HALDOIback to textback to text
- 19 articleLong-term treatment effects in chronic myeloid leukemia.Journal of Mathematical Biology2017, 1-26HALDOIback to text
- 20 articleImplication of the Autologous Immune System in BCR-ABL Transcript Variations in Chronic Myelogenous Leukemia Patients Treated with Imatinib..Cancer Research7519October 2015, 4053-62HALDOIback to text
- 21 articleIdentification of Nascent Memory CD8 T Cells and Modeling of Their Ontogeny.Cell Systems43February 2017, 306-317HALDOIback to textback to text
- 22 articleInferring gene regulatory networks from single-cell data: a mechanistic approach.BMC Systems Biology111November 2017, 105HALDOIback to text