

Section: Scientific Foundations
Models: Development and reduction of models of bacterial regulatory networks
Participants : Sara Berthoumieux, Eugenio Cinquemani, Jérôme Izard, Johannes Geiselmann, Hidde de Jong, Stéphane Pinhal, Delphine Ropers [Correspondent] , Valentin Zulkower.
The adaptation of bacteria to changes in their environment is controlled on the molecular level by large and complex interaction networks involving genes, mRNAs, proteins, and metabolites (Figure 2 ). The elucidation of the structure of these networks has much progressed as a result of decades of work in genetics, biochemistry, and molecular biology. Most of the time, however, it is not well understood how the response of a bacterium to a particular environmental stress emerges from the interactions between the molecular components of the network. This has called forth an increasing interest in the mathematical modeling of the dynamics of biological networks, in the context of a broader movement called systems biology.
|
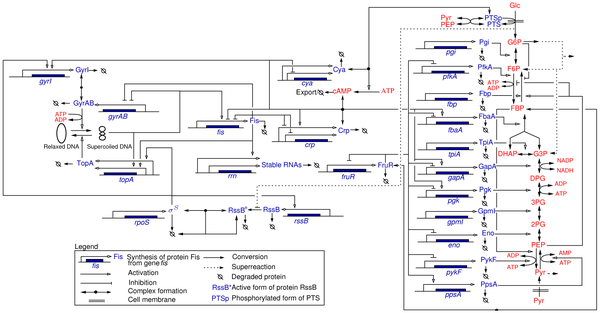
In theory, it is possible to write down mathematical models of biochemical networks, and study these by means of classical analysis and simulation tools. In practice, this is not easy to achieve though, as quantitative data on kinetic parameters are usually absent for most systems of biological interest. Moreover, the models include a large number of variables, are strongly nonlinear and include different time-scales, which make them difficult to handle both mathematically and computationally. A possible approach to this problem has been to use approximate models that preserve essential dynamical properties of the networks. Different approaches have been proposed in the literature, such as the use of approximations of the typical response functions found in gene and metabolic regulation and the reduction of the model dimension by decomposing the system into fast and slow subsystems. These reductions and approximations result in simplified models that are easier to analyze mathematically and for which parameter values can be more reliably estimated from the available experimental data.
Model reduction approaches are exploited in IBIS to gain a better understanding of the ability of E. coli to adapt to a various nutritional and other environmental stresses, such as carbon, phosphate, and nitrogen starvation. We are particularly interested in gaining a better understanding of the role of the so-called global regulators of gene expression in shaping the adaptive response of the bacteria. Moreover, we study the interactions between metabolism and gene expression in the adaptation of E. coli to changes in available carbon sources. These topics are studied in collaboration with the BAMBOO and COMORE project-teams at INRIA.