Section: New Results
Interactive quantum chemistry
Participants : Mael Bosson, Caroline Richard, Antoine Plet, Sergei Grudinin, Stephane Redon.
Interactive simulation tools allow users to take advantage of their knowledge and intuition to understand physical properties and prototype new devices. To accurately describe bond breaking, bond formation, charge transfer or other electronic phenomena, interactive simulation should ideally be based on quantum mechanics. However, solving quantum chemistry models at interactive rates is a challenging task. Thanks to the algorithms developed in the group, SAMSON is the first software to propose interactive quantum chemistry.
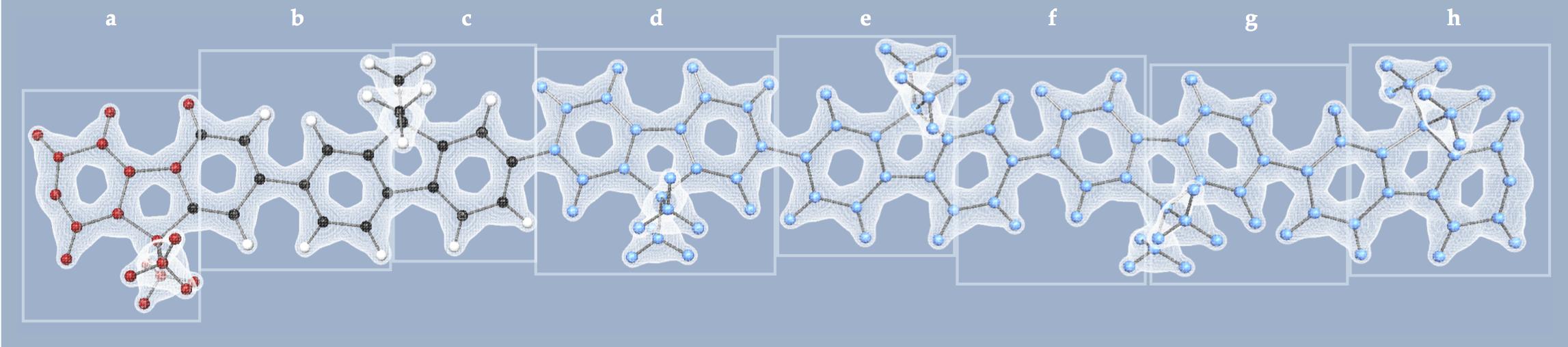
A first contribution allows for interactive quantum chemistry with systems up to a few hundred atoms [6] . The method is based on a divide-and-conquer (D&C) approach. The D&C technique subdivides the system into many subsystems (a–h on the Figure 10 ). Each of them involves a diagonalisation at each time step. To treat larger systems, we introduce a new algorithm: Block-Adaptive Quantum Mechanics (BAQM) [5] from the combination of two new components.
Block-adaptive Cartesian mechanics
By freezing atomic positions in some subsystems (d–h on the Figure 10 ) (with atoms in blue), we may avoid updating some eigenproblems. The Block-adaptive Cartesian mechanics component takes advantage of this to control the simulation cost by adaptively adjusting the number of diagonalisations, based on the forces applied to the atoms. Only the subsystems with the largest applied forces are allowed to have mobile atoms.
Adaptive reduced-basis quantum mechanics
Solving even just one of the subsystem's eigenproblem may be too costly to achieve interactive rates. The Adaptive reduced-basis quantum mechanics component projects the equation in an adaptive reduced basis composed of low-energy eigenvectors that have been computed at a previous time step, to benefit from temporal coherence between successive eigenproblems (subsystems (b) and (c) with atoms in black and white on the Figure 10 ). We use a simple distance to decide on the fly when to automatically update the reduced basis during the simulation (subsystem (a) with atoms in red on the Figure 10 ).
We demonstrated that BAQM may accelerate geometry optimization for several atomic systems. Indeed, each step is solved significantly faster by constraining some nuclei and electrons, and, by focusing computational resources on the most active parts of the system, we obtain a faster potential energy descent. The proposed BAQM approach also allows for interactive rates with many atomic systems.