

Section: Research Program
RNA
At the secondary structure level, we contributed novel generic techniques applicable to dynamic programming and statistical sampling, and applied them to design novel efficient algorithms for probing the conformational space. Another originality of our approach is that we cover a wide range of scales for RNA structure representation. For each scale (atomic, sequence, secondary and tertiary structure...) cutting-edge algorithmic strategies and accurate and efficient tools have been developed or are under development. This offers a new view on the complexity of RNA structure and function that will certainly provide valuable insights for biological studies.
3D modeling was supported by the Digiteo project Japarin-3D . Statistical potentials were supported by Carnage and Itsnap .
Dynamic programming and complexity
Participants : Alain Denise, Yann Ponty, Antoine Soulé.
Common activity with J. Waldispühl (McGill).
Ever since the seminal work of Zuker and Stiegler, the field of RNA bioinformatics has been characterized by a strong emphasis on the secondary structure. This discrete abstraction of the 3D conformation of RNA has paved the way for a development of quantitative approaches in RNA computational biology, revealing unexpected connections between combinatorics and molecular biology. Using our strong background in enumerative combinatorics, we propose generic and efficient algorithms, both for sampling and counting structures using dynamic programming. These general techniques have been applied to study the sequence-structure relationship [77] , the correction of pyrosequencing errors [71] , and the efficient detection of multi-stable RNAs (riboswitches) [72] , [73] .
|
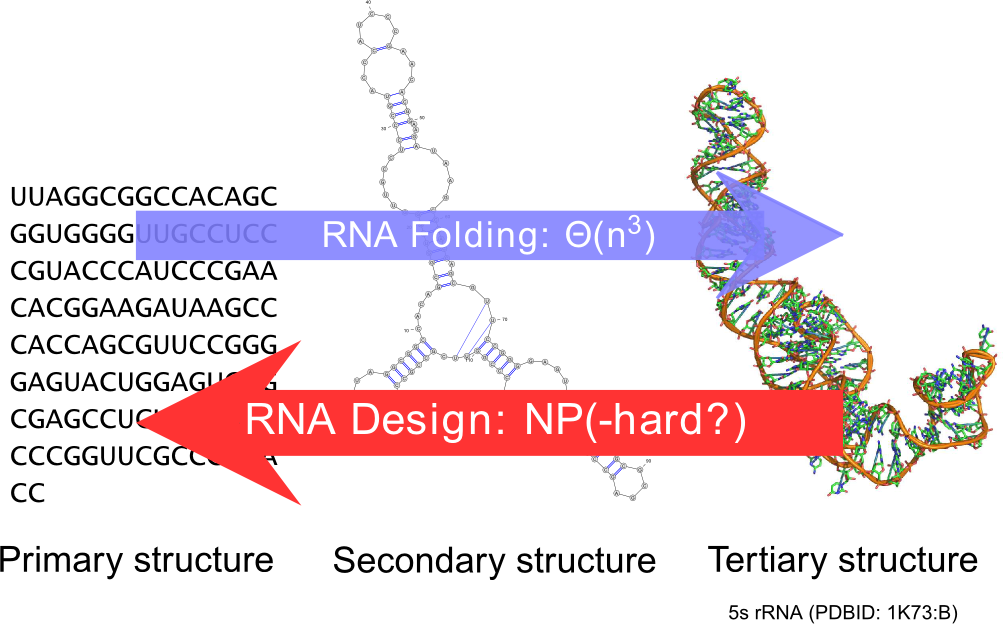
RNA design.
Participants : Alain Denise, Vincent Le Gallic, Yann Ponty.
Joint project with S. Vialette (Marne-la-Vallée), J. Waldispühl (McGill) and Y. Zhang (Wuhan).
It is a natural pursue to build on our understanding of the secondary structure to construct artificial RNAs performing predetermined functions, ultimately targeting therapeutic and synthetic biology applications. Towards this goal, a key element is the design of RNA sequences that fold into a predetermined secondary structure, according to established energy models (inverse-folding problem). Quite surprisingly, and despite two decades of studies of the problem, the computational complexity of the inverse-folding problem is currently unknown.
Within our group, we offer a new methodology, based on weighted random generation [54] and multidimensional Boltzmann sampling, for this problem. Initially lifting the constraint of folding back into the target structure, we explored the random generation of sequences that are compatible with the target, using a probability distribution which favors exponentially sequences of high affinity towards the target. A simple posterior rejection step selects sequences that effectively fold back into the latter, resulting in a global sampling pipeline that showed comparable performances to its competitors based on local search [60] .
Towards 3D modeling of large molecules
Participants : Alain Denise, Mélanie Boudard.
Joint project with D. Barth (Versailles) and J. Cohen (Paris-Sud).
The modeling of large RNA 3D structures, that is predicting the three-dimensional structure of a given RNA sequence, relies on two complementary approaches. The approach by homology is used when the structure of a sequence homologous to the sequence of interest has already been resolved experimentally. The main problem then is to calculate an alignment between the known structure and the sequence. The ab initio approach is required when no homologous structure is known for the sequence of interest (or for some parts of it). We work in both directions.
Statistical and robotics-inspired models for structure and dynamics
Participants : Julie Bernauer, Rasmus Fonseca.
Despite being able to correctly model small globular proteins, the computational structural biology community still craves for efficient force fields and scoring functions for prediction but also good sampling and dynamics strategies.
Our current and future efforts towards knowledge-based scoring function and ion location prediction have been described in 3.1.4 .
Over the last two decades a strong connection between robotics and computational structural biology has emerged, in which internal coordinates of proteins are interpreted as a kinematic linkage with rotatable bonds as joints and corresponding groups of atoms as links [76] , [51] , [64] , [63] . Initially, fragments in proteins limited to tens of residues were modeled as a kinematic linkage, but this approach has been extended to encompass (multi-domain) proteins [62] . For RNA, progress in this direction has been realized as well. A kinematics-based conformational sampling algorithm, KGS, for loops was recently developed [58] , but it does not fully utilize the potential of a kinematic model. It breaks and recloses loops using six torsional degrees of freedom, which results in a finite number of solutions. The discrete nature of the solution set in the conformational space makes difficult an optimization of a target function with a gradient descent method. Our methods overcome this limitation by performing a conformational sampling and optimization in a co-dimension 6 subspace. Fragments remain closed, but these methods are limited to proteins. Our objective is to extend the approach proposed in [58] , [76] to nucleic acids and protein/nucleic acid complexes with a view towards improving structure determination of nucleic acids and their complexes and in silico docking experiments of protein/RNA complexes. For that purpose, we have developed a generic strategy for differentiable statistical potentials [2] , [74] that can be directly integrated in the procedure.
Results from in silico docking experiments will also directly benefit structure determination of complexes which, in turn, will provide structural insights in nucleic acid and protein/nucleic acid complexes. From the small proof-of-concept single chain protein implementation of the KGS strategy, we have developed a robust preliminary implementation that can handle RNA and will be further developed to account for multi-chain molecules. Rasmus Fonseca, post-doctoral scholar in the project is currently performing an extensive computational and biological validation.