

Section: New Results
Meristem functioning and development
In axis 2 work focuses on the creation of a virtual meristem, at cell resolution, able to integrate the recent results in developmental biology and to simulate the feedback loops between physiology and growth. The approach is subdivided into several sub-areas of research.
Data acquisition and design of meristem models
-
Improvement of the MARS-ALT pipeline robustness.
Meristem, laser microscopy, image reconstruction, cell segmentation, automatic lineaging
Participants : Léo Guignard,, Christophe Godin, Christophe Pradal, Grégoire Malandain [Morpheme, Inria] , Gaël Michelin [Morpheme, IPL Morphogenetics, Inria] , Guillaume Baty, Sophie Ribes [IBC, UM] , Jan Traas [RDP, ENS Lyon] , Patrick Lemaire [CRBM, CNRS] , Yassin Refahi [RDP, ENS-Lyon / Sainsbury Lab, Cambridge, UK] .
This research theme is supported by a PhD FRM grant, Jan Traas's ERC, Inria ADT programme and the Morphogenetics Inria Project Lab.
The MARS-ALT (Multi-Angles Registration and Segmentation - Automatic Lineage Tracking) software pipeline [5] automatically performs a segmentation at cell resolution from 3D or 2D voxel images where the membranes/walls are marked (by a die for example) and makes it possible to follow the lineage of these cells through time.
This year, the ALT tracking pipeline has been reformulated by using a generic cell modeling approach (enabling for example more than one cell division), and both stability and robustness were improved. The modeling approach is generic and can be used on other kind of data (nuclei, human cells, ...). Moreover, the architecture of the image processing components has been modified (plugin approach) and integrated with the TissueLab platform. The new segmentation-tracking library is called TimageTK will be released at the beginning of next year.
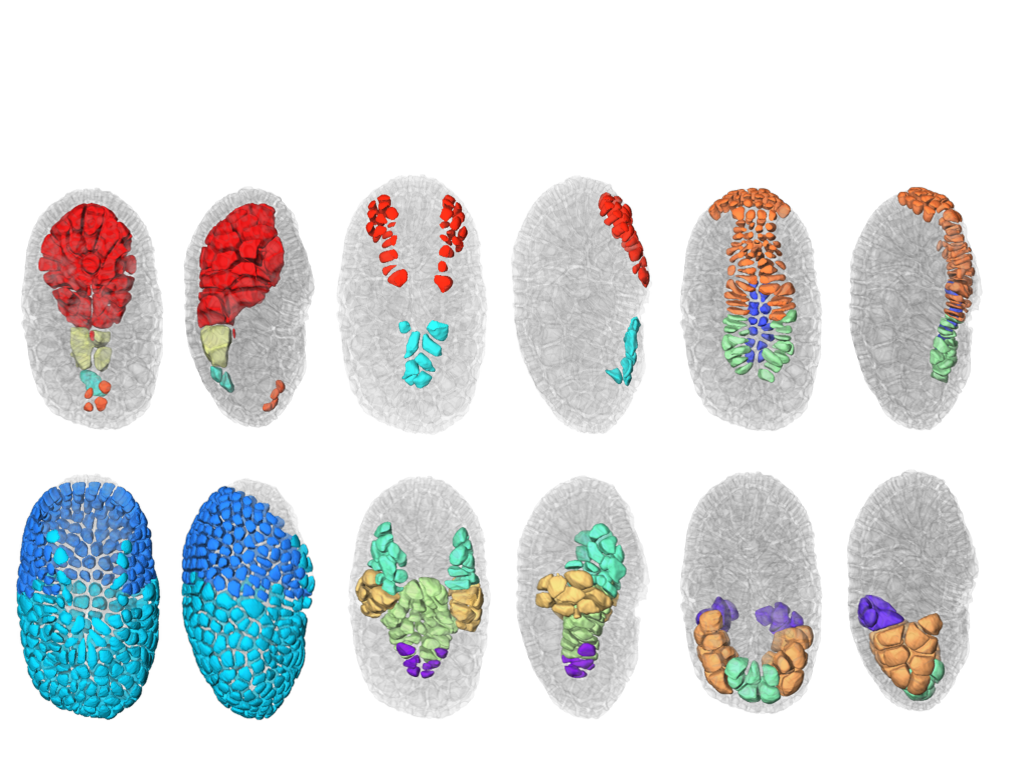
We also finalize the development of a a new segmentation and tracking pipeline, ASTEC (Adaptive Segmentation and Tracking of Embryonic Cells). ASTEC is a one-pass algorithm (in contrast to MARS-ALT, that perform first the segmentation and then the tracking in two-passes) that is best suited for movies with numerous close time-points acquired at high spatio-temporal resolution. This pipeline takes advantage of information redundancy across the movies and biological knowledge on the segmented organism to constrain and improve the segmentation and the tracking. We used this one-pass algorithm to segment and track all cell shapes of a developing embryo of the marine invertebrate Phallusia mammillata. As a result we obtained the full track of the shapes of all the cells from the 64 cell stage up to the early tailbud stage (1030 cells undergoing 640 division events followed across 180 time-points through 6 hours of development imaged every 2 minutes, Figure 2).
Based on this quantitative digital representation, we systematically identified cell fate specification events up to the late gastrula stage. Computational simulations revealed that remarkably simple rules integrating measured cell-cell contact areas with spatio-temporal expression data for extracellular signalling molecules are sufficient to explain most early cell inductions. This work suggests that in embryos developing with stereotyped cell shapes and positions (like Phallusia mammillata embryos), the genomic constraints for precise gene expression levels are relaxed, thereby allowing rapid genome evolution.
-
Creating mesh representation of cellular structures.
Participants : Guillaume Cerutti, Sophie Ribes, Christophe Godin, Géraldine Brunoud [RDP, ENS] , Carlos Galvan-Ampudia [RDP, ENS] , Teva Vernoux [RDP, ENS] , Yassin Refahi [RDP, ENS, Sainsbury Lab] .
This research theme is supported the HFSP project Biosensors.
To produce a more efficient data structure accounting for the geometry of cellular tissues, we studied the problem of reconstructing a mesh representation of cells in a complex, multi-layered tissue structure, based either on membrane/wall images segmented using MARS or on nuclei images of shoot apical meristems. The construction of such mesh structures for plant tissues is currently a missing step in the existing image analysis pipelines.
We developed tools to reconstruct a 3D cell complex representing the tissue, based on the dual simplicial complex of cell adjacencies. This set of tetrahedra is optimized from a reasonable initial guess to match the adjacencies in the tissue, which proved to produce a very faithful reconstruction [62]. We also developed a set of methods to triangulate such reconstructions, and enhance the quality of triangular mesh representations of plant tissue, simultaneously along several criteria [61].
These tools are integrated in the DRACO-STEM computational pipeline released as an independent package to enable biomechanical simulations on real-world data.
-
Design of 3D digital atlases of tissue development.
Participants : Sophie Ribes, Yassin Refahi [RDP, ENS, Sainsbury Lab] , Guillaume Cerutti, Christophe Godin, Christophe Pradal, Christophe Pradal, Frédéric Boudon, Gregoire Malandain [RDP, ENS] , Gaël Michelin [RDP, ENS] , Jan Traas [RDP, ENS] , Teva Vernoux [RDP, ENS] , Patrick Lemaire [CRBM, CNRS] .
This research theme is supported the Inria Project Lab Morphogenetics, the ADT Mars-Alt and the HFSP project Biosensors.
To organize the various genetic, physiological, physical, temporal and positional informations, we build a spatialized and dynamic database [72]. This database makes it possible to store all the collected information on a virtual 3D structure representing a typical organ. Each piece of information has to be located spatially and temporally in the database. Tools to visually retrieve and manipulate the information, quantitatively through space and time are being developed. For this, the 3D structure of a typical organ has been created at the different stages of development of the flower bud. This virtual structure contains spatial and temporal information on mean cell numbers, cell size, cell lineages, possible cell polarization (transporters, microtubules), and gene expression patterns. Such 3D digital atlas is mainly descriptive. However, like for classical databases, specific tools make it possible to explore the digital atlas according to main index keys, in particular spatial and temporal keys. Both a dedicated language and a 3D user interface are being designed to investigate and query the 3D virtual atlas. Current developments of this tool consist in using directly the segmented images produced from laser microscopy to build the atlas. To better represent the development of a biological population, a method to compute an "average" structure is being investigated.

Shape analysis of meristems
Participants : Jonathan Legrand, Guillaume Cerutti, Pierre Fernique, Frédéric Boudon, Yann Guédon, Christophe Godin, Pradeep Das [RDP, ENS] , Arezki Boudaoud [RDP, ENS] .
The MARS-ALT pipeline provides rich spatio-temporal data sets for analyzing the development of meristems, since it allows to performs 3D cell-segmentation and to compute cell-lineage. This enable the extraction and study of spatio-temporal properties of a tissue at cellular scale. To facilitate the analysis and to structure the obtained data have implemented a dedicated temporal graph structure. In this graph, vertex are cells and edges are spatial or temporal relationships, thus proposing a natural representation of the growing tissue. Various variables can be attached either to the vertices (e.g. cell volume, inertia axes) or the edges (e.g. wall surface, distance between cell centroids). This graph may be augmented by new variables resulting from various spatial or temporal filtering (e.g. cell volumetric growth). Looking at homogeneous regions in the variable space, cellular patterns can be identified, by clustering methods for instance.
Considering the highly-structured nature of our data (time and space structuring) and the potential diversity and heterogeneity of possible cell descriptors, we developed two complementary approaches:
-
A first one that favours the spatial structuring: In this approach, the cell neighbourhood and the cell descriptors are jointly taken into account in a clustering approach whose objective is to identify a small number of clusters corresponding to well-defined cell identities. Once the cells have been labelled using the clustering algorithm, cell generation distributions may be estimated on the basis of the labelled lineage trees.
-
A second one that favours the temporal structuring: In this approach, the data of interest are lineage forest and the only spatial structuring taken into account corresponds to siblings with respect to a given parent cell. In a first step, cell identities are inferred on the basis of the cell descriptors taking into account lineage relationships using hidden Markov tree models and the spatial regions that emerge from the cell identity labelling are then characterized. This second approach is supported by the fact that cell topology is only affected by division which makes highly relevant the local spatial information taken into account in this approach.
Mechanical models of plant tissues
Participants : Jean-Philippe Bernard, Olivier Ali, Christophe Godin, Benjamin Gilles, Frédéric Boudon, Ibrahim Cheddadi, Jan Traas [ENS-Lyon] , Olivier Hamant [ENS-Lyon] , Arezki Boudaoud [ENS-Lyon] .
This research theme is supported by the Inria Project Lab Morphogenetics and Jan Traas's ERC.
During the previous years, we set up a mechanical model of a growing shoot apical meristem (the specific tissue at the very tip of plants where stem cells are active and produce new organs such as branches, leafs and flowers). The aim of this project is to provide a computational framework for simulating growth of multicellular plant tissue. This framework integrates a theoretical description of the major biophysical processes at stake. A first version of the model, based on a static description of the tissues rheological properties, has been published last year [57].
This year, we used this model in close collaboration with biologists to investigate the coupling between growth and cell wall remodeling required in early stages of organogenesis. Our simulations pointed out that cell wall remodelling and growth initiation have to be co-regulated in order to initiate young organs formation. Biologists unraveled a biochemical signaling pathway that could explain this synergy. This joint work has been submitted to a high factor Biology journal.
In parallel, we also improved the underlying biophysical theory. One important aspect of the problem is the multiscale interconnections between mechanical forces generated at the scale of the whole tissue and the molecular response to these forces at the subcellular level. To tackle this issue, we established a parcimonous molecular description of the cell wall (one of the main organelle involded in growth) attesting for its biochemical behavior under mechanical loading. This description has been formalized as a unidimensional toy-model. With this toy-model we exposed how large-scale behavior of an expanding cell wall could be controlled by the biochemical behavior of a limited set of molecular actors. This work has been published [14].
Additionally, we started to work on the integration of a feedback loop between mechanical stresses and growth (PhD work of Hadrien Oliveri started in Oct. 2015). A close study of this feedback mechanism made us refine several aspects of our modelling approach. On the molecular scale, we introduced a tensor formalism to quantify cell polarity, based on the description of its cortical microtubule network. Microtubules being stress-sensitive, we described this feedback loop through the coupling between this polarity tensor and the mechanical stress field. In parallel, through a parcimonous model of microtubule-guided cell wall turnover, we derived an expression of the stiffness tensor as a function of cell polarity. This enabled us to relate subcellular stress-induced dynamics of microtubules to the evolution of large scale rheological properties of the tissue. We also started to work on the numerical implementation of this feedback mechanism. FEM-based simulations have been carried out on simple structures as proof of concept. By doing so we assessed the numerical valididy of our resolution scheme along with the relevance of our biophysical description.
Mechanical modelling of embryo morphogenesis.
Participants : Bruno Leggio, Emmanuel Faure, Patrick Lemaire [CRBM, CNRS] , Christophe Godin.
A work on data analysis and modelling of morphogenesis and development in embryos of ascidians has been started this year. It comprises two main branches: starting from segmented data at cellular resolution, global and local symmetries of embryo development were analyzed. An analysis in terms of entropy of conserved embryonic properties was developed in order to characterise different stages of development as well as different tissues.
In parallel, a mechanical and topological analysis of cell-cell interactions was carried out. This lead us to develop a new and original physical model of cleavage-plane determination in different tissues, with the goal of understanding the role of purely mechanical interactions in shaping ascidian embryos.
Modelling the influence of dimerisation sequence dissimilarities on the auxin signalling network
Participants : Jonathan Legrand, Yann Guédon, Jean-Benoist Léger [INRA, MIA, Paris] , Stéphane Robin [INRA, MIA, Paris] , Teva Vernoux [ENS-Lyon] .
Auxin is a major phytohormone involved in many developmental processes by controlling gene expression through a network of transcriptional regulators. In Arabidopsis thaliana, the auxin signalling network is made of 52 potentially interacting transcriptional regulators, activating or repressing gene expression. All the possible interactions were tested in two-way yeast-2-hybrid experiments. Our objective was to characterise this auxin signalling network and to quantify the influence of the dimerisation sequence dissimilarities on the interaction between transcriptional regulators [20]. We applied model-based graph clustering methods relying on connectivity profiles between transcriptional regulators. Incorporating dimerisation sequence dissimilarities as explanatory variables, we modelled their influence on the auxin network topology using mixture of linear models for random graphs. Our results provide evidence that the network can be simplified into four groups, three of them being closely related to biological groups. We found that these groups behave differently, depending on their dimerisation sequence dissimilarities, and that the two dimerisation sub-domains might play different roles. We propose here the first pipeline of statistical methods combining yeast-2-hybrid data and protein sequence dissimilarities for analysing protein-protein interactions. We unveil using this pipeline of analysis the transcriptional regulator interaction modes.
Model integration
Participants : Frédéric Boudon, Christophe Godin, Guillaume Cerutti, Jean-Louis Dinh, Eugenio Azpeitia, Jan Traas.
This research theme is supported by the Morphogenetics Inria Project Lab.
One key aspect of our approach is the development of a computer platform dedicated to programming virtual tissue development, TissueLab. This platform, based on OpenAlea, will be used to carry out integration of the different models developed in this research axis. In the past year, progress has been made in defining a generic tissue data structure that would be visualized, manipulated and updated through this platform. Currently, robust geometric operations such as division are implemented and tested. Moreover, a redesign of the structure based on more elaborated formalisms such as combinatorial maps is being investigated. A 2D version is being developed in the context of Jean-Louis's Dinh PhD thesis, and will be described in a forthcoming book chapter.