

Section: Research Program
Inference of gene regulatory networks from time-series data
Participants : Eugenio Cinquemani [Correspondent] , Johannes Geiselmann, Hidde de Jong, Stephan Lacour, Aline Marguet, Michel Page, Corinne Pinel, Delphine Ropers.
Measurements of the transcriptome of a bacterial cell by means of DNA microarrays, RNA sequencing, and other technologies have yielded huge amounts of data on the state of the transcriptional program in different growth conditions and genetic backgrounds, across different time-points in an experiment. The information on the time-varying state of the cell thus obtained has fueled the development of methods for inferring regulatory interactions between genes. In essence, these methods try to explain the observed variation in the activity of one gene in terms of the variation in activity of other genes. A large number of inference methods have been proposed in the literature and have been successful in a variety of applications, although a number of difficult problems remain.
Current reporter gene technologies, based on Green Fluorescent Proteins (GFPs) and other fluorescent and luminescent reporter proteins, provide an excellent means to measure the activity of a gene in vivo and in real time (Figure 3). The underlying principle of the technology is to fuse the promoter region and possibly (part of) the coding region of a gene of interest to a reporter gene. The expression of the reporter gene generates a visible signal (fluorescence or luminescence) that is easy to capture and reflects the expression of a gene of interest. The interest of the reporter systems is further enhanced when they are applied in mutant strains or combined with expression vectors that allow the controlled induction of any particular gene, or the degradation of its product, at a precise moment during the time-course of the experiment. This makes it possible to perturb the network dynamics in a variety of ways, thus obtaining precious information for network inference.
|
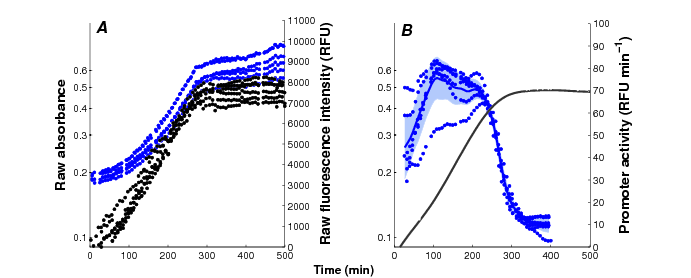
The specific niche of IBIS in the field of network inference has been the development and application of genome engineering techniques for constructing the reporter and perturbation systems described above, as well as the use of reporter gene data for the reconstruction of gene regulation functions. We have developed an experimental pipeline that resolves most technical difficulties in the generation of reproducible time-series measurements on the population level. The pipeline comes with data analysis software that converts the primary data into measurements of time-varying promoter activities. In addition, for measuring gene expression on the single-cell level by means of microfluidics and time-lapse fluorescence microscopy, we have established collaborations with groups in Grenoble and Paris. The data thus obtained can be exploited for the structural and parametric identification of gene regulatory networks, for which methods with a solid mathematical foundation are developed, in collaboration with colleagues at ETH Zürich and EPF Lausanne (Switzerland). The vertical integration of the network inference process, from the construction of the biological material to the data analysis and inference methods, has the advantage that it allows the experimental design to be precisely tuned to the identification requirements.