
Keywords
Computer Science and Digital Science
- A3.1. Data
- A3.2. Knowledge
- A3.3.2. Data mining
- A3.3.3. Big data analysis
- A3.4. Machine learning and statistics
- A6.1. Methods in mathematical modeling
- A6.2. Scientific computing, Numerical Analysis & Optimization
Other Research Topics and Application Domains
- B1.1.7. Bioinformatics
- B1.1.10. Systems and synthetic biology
- B3. Environment and planet
1 Team members, visitors, external collaborators
Research Scientists
- David Sherman [Team leader, Inria, Senior Researcher, HDR]
- Pascal Durrens [CNRS, Researcher, HDR]
- Alain Franc [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, Senior Researcher, HDR]
- Clémence Frioux [Inria, Researcher]
PhD Students
- Mohamed Anwar Abouabdallah [Inria]
- Maxime Lecomte [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, from Nov 2020]
Technical Staff
- Ariane Badoual [Inria, Engineer, from Oct 2020]
- Philippe Chaumeil [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, Engineer]
- Jean-Marc Frigerio [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, Engineer]
- Franck Salin [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, Engineer]
Interns and Apprentices
- Ariane Badoual [Inria, from Feb 2020 until Jul 2020]
- Maxime Lecomte [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement, from Feb 2020 until Aug 2020]
Administrative Assistants
- Catherine Cattaert Megrat [Inria, until Oct 2020]
- Roweida Mansour El Handawi [Inria, from Oct 2020]
External Collaborator
- Simon Labarthe [Institut national de recherche pour l'agriculture, l'alimentation et l'environnement]
2 Overall objectives
Diversity, evolution, and inheritance form the heart of modern biological thought. Modeling the complexity of biological systems has been a challenge of theoretical biology for over a century 43 and flourished with the evolution of data for describing biological diversity, most recently with the transformative development of high-throughput sequencing. However, most concepts and tools in ecology and population genetics for capitalizing on this wealth of data are still not adapted to high throughput data production. A better connection between high-throughput data production and tool evolution is highly needed: computational biodiversity.
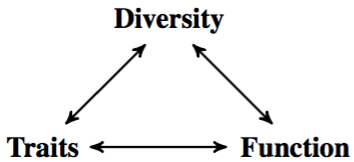
Paradoxically, diversity emphasizes differences between biological objects, while modeling aims at unifying them under a common framework. This means that there is a limit beyond which some components of diversity cannot be mastered by modeling. We need efficient methods for recognizing patterns in diversity, and linking them to patterns in function. It is important to realize that diversity in function is not the same as coupling observed diversity with function. Diversity informs both the study of traits, and the study of biological functions (Figure 1). The double challenge is to measure these links quickly and precisely with pattern recognition, and to explore the relations between diversity in traits and diversity in function through modeling
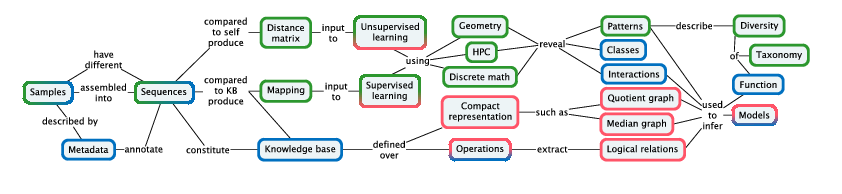
Pleiade links recognition of patterns, classes, and interactions with applications in biodiversity studies and biotechnology. We develop distance methods for NGS datasets at different levels of organization: between genomes, between individual organisms, and between communities; and develop high-performance pattern recognition and statistical learning techniques for analyzing the resulting point clouds. We refine inferential methods for building hierarchical models of networks of cellular functions, exploiting the mathematical relations that are revealed by large-scale comparison of related genomes and their models. We combine these methods into integrated e-Science solutions to place these tools directly in the hands of biologists.
Our methodology (Figure 2) is designed pragmatically to advance the state of the art in applications from biodiversity and biotechnology: molecular based systematics and community ecology, annotation and modeling for biotechnology.
3 Research program
3.1 A Geometric View of Diversity
Diversity may be studied as a set of dissimilarities between objects. The underlying mathematical construction is the notion of distance. Knowing a set of objects, it is possible, after computation of pairwise distances, or sometimes dissimilarities, to build a Euclidean image of it as a point cloud in a space of relevant dimension. Then, diversity can be associated with the shape of the point cloud. The human eye is often far better than an algorithm at recognizing a pattern or shape. One objective of our project is to narrow the gap between the story that a human eye can tell, and that an algorithm can tell. Several directions will be explored. First, this requires mastering classical tools in dimension reduction, mainly algebraic tools (PCA, NGS, Isomap, eigenmaps, etc ...). Second, neighborhoods in point clouds naturally lead to graphs describing the neighborhood networks. There is a natural link between modular structures in distance arrays and communities on graphs. Third, points (representing, say, DNA sequences) are samples of diversity. Dimension reduction may show that they live on a given manifold. This leads to geometry (differential or Riemannian geometry). It is expected that some properties of the manifold can tell something of the constraints on the space where measured individuals live. The connection between Riemannian geometry and graphs, where weighted graphs are seen as mesh embedded in a manifold, is currently an active field of research 42, 41. See as well 44 for a link between geometric structure, linear and nonlinear dimensionality reduction.
Biodiversity and high-performance computing: Most methods and tools for characterizing diversity have been designed for datasets that can be analyzed on a laptop, but NGS datasets produced for metabarcoding are far too large. Data analysis algorithms and tools must be revisited and scaled up. We will mobilize both distributed algorithms like the Arnoldi method and new algorithms, like random projection or column selection methods, to build point clouds in Euclidean spaces from massive data sets, and thus to overcome the cubic complexity of computation of eigenvectors and eigenvalues of very large dense matrices. We will also link distance geometry 35 with convex optimization procedures through matrix completion 24, 26.
Intercalibration: There is a considerable difference between supervised and unsupervised clustering: in supervised clustering, the result for an item is independent from the result for an item , whereas in unsupervised clustering, the result for an item (e.g. the cluster it belongs to, and its composition) depends on nearby items . Which means that the result may change if some items are added to or subtracted from the sample. This raises the more global problem of how to merge two studies to yield a more comprehensive view of biodiversity?
See 12 for some of our recent work linking the distance geometry problem, nonlinear mapping, and weighted least-squares scaling.
3.2 Knowledge Management for Biology
The heterogenous data generated in computational molecular biology and ecology are distinguished not only by their volume, but by the richness of the many levels of interpretation that biologists create. The same nucleic acid sequence can be seen as a molecule with a structure, a sequence of base pairs, a collection of genes, an allele, or a molecular fingerprint. To extract the maximum benefit from this treasure trove we must organize the knowledge in ways that facilitate extraction, analysis, and inference. Our focus has been on the efficient representation of relations between biological objects and operations on those representations, in particular heuristic analyses and logical inference.
Pleiade will develop applications in comparative genomics of related organisms, using new mathematical tools for representing compactly, at different scales of difference, comparisons between related genomes. New methods based on distance geometry will refine these comparisons. Compact representations can be stored, exchanged, and combined. They will form the basis of new simultaneous genome annotation methods, linked directly to abductive inference methods for building functional models of the organisms and their communities.
Since a goal of Pleiade is to integrate diversity throughout the analysis process, it is necessary to incorporate diversity as a form of knowledge that can be stored in a knowledge base. Diversity can be represented using various compact representations, such as trees and quotient graphs storing nested sets of relations. Extracting structured representations and logical relations from integrated knowledge bases (Figure 2) will require domain-specific query methods that can express forms of diversity.
3.3 Community-scale metabolic modeling
The emergent metabolism of microbial communities can be qualitatively modeled using a boolean approximation of metabolic dynamics13. In this approach the behavior of the system is described by logical rules that activate a given reaction as soon as its substrates become available; numerical parameters such as stochiometry or enzyme kinetics are ignored in favor of graph topology and paths. The advantage is that such qualitative models, unlike quantitative methods such as flux balance analysis, do not require the assumption that the system is stationary and can model systems where cells are constantly growing or constantly reproducing.
Network expansion, introduced in 29 as a recursive traversal of the structure of a metabolic graph, lends itself to concise definition using answer set programming33 and thus to efficient implementation using SAT solvers 32. In practice, using ASP for metabolic modeling makes it possibe to define both the activation of metabolic reactions in different conditions, and the constraints and optimizations needed to find solutions in a combinatorically large state space.
We focus in particular on the key question of determining minimal communities, subsets of the organisms present in an environment that are sufficient to produce a chosen behavior 30. The methodological goal here is to identify key species in a community through use of ASP to rapidly explore the state space and thus, through heuristic resolution of combinatorial problems, provide the guarantees an exhaustive search with a greatly reduced computational cost 9.
3.4 Modeling by successive refinement
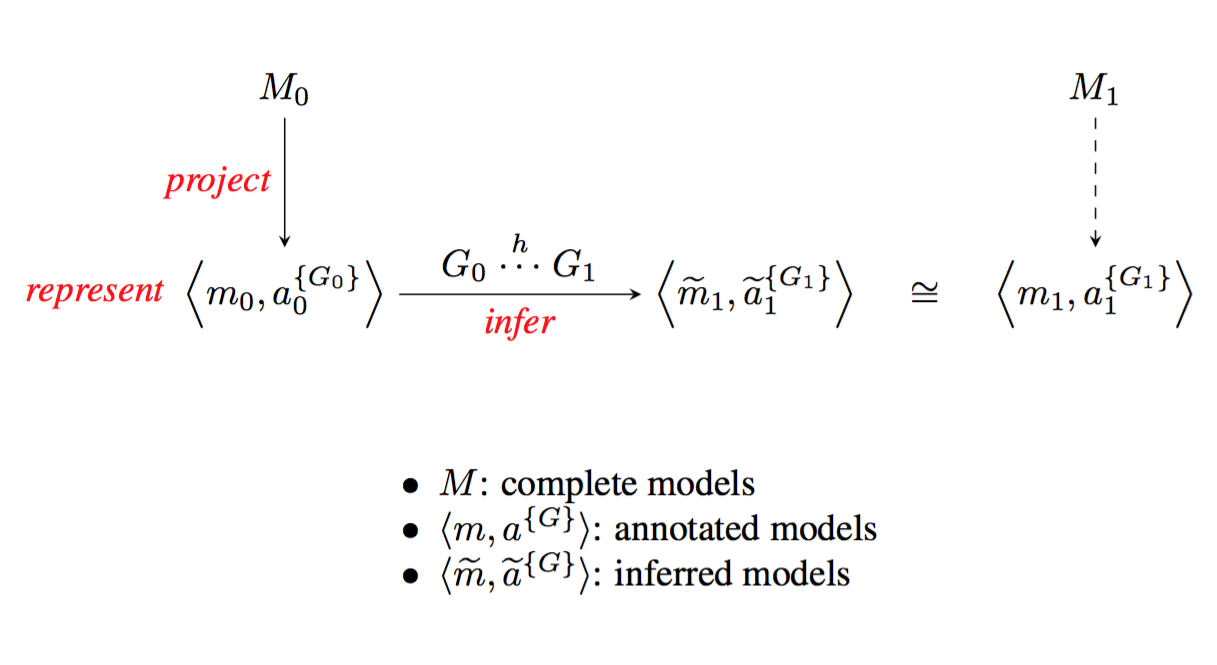
Describing the links between diversity in traits and diversity in function will require comprehensive models, assembled from and refining existing models. A first level of refinement is inferring a new model for a specific organism, on the basis of an annotated projection and knowledge of genome-to-genome relations (figure 4).
Beyond that, a recurring difficulty in building comprehensive models of biological systems is that accurate models for subsystems are built using different formalisms and simulation techniques, and hand-tuned models tend to be so focused in scope that it is difficult to repurpose them 22. Our belief is that a sustainable effort in building efficient behavioral models must proceed incrementally, rather than by modeling individual processes de novo. Hierarchical modeling19 is one way of combining specific models into networks. Effective use of hierarchical models requires both formal definition of the semantics of such composition, and efficient simulation tools for exploring the large space of complex behaviors. We have previously shown that this approach can be effective for certains kinds of systems in biotechnology 2, 23 and medicine 21. Our challenge is to adapt incremental, hierarchical refinement to modeling organisms and communities in metagenomic and comparative genomic applications.
4 Application domains
4.1 Genome and transcriptome annotation, to model function
Sequencing genomes and transcriptomes provides a picture of how a biological system can function, or does function under a given physiological condition. Simultaneous sequencing of a group of related organisms is now a routine procedure in biological laboratories for studying a behavior of interest, and provides a marvelous opportunity for building a comprehensive knowledge base of the relations between genomes1, 6. Key elements in mining these relations are: classifying the genes in related organisms and the reactions in their metabolic networks, recognizing the patterns that describe shared features, and highlighting specific differences.
Pleiade will develops applications in comparative genomics of related organisms, using new mathematical tools for representing compactly, at different scales of difference, comparisons between related genomes. New methods based on computational geometry refine these comparisons. Compact representations can be stored, exchanged, and combined. They will form the basis of new simultaneous genome annotation methods, linked directly to abductive inference methods for building functional models of the organisms and their communities.
Our ambition in biotechnology is to permit the design of synthetic or genetically selected organisms at an abstract level, and guide the modification or assembly of a new genome. Our effort is focused on two main applications: genetic engineering and synthetic biology of oil-producing organisms (biofuels in CAER, palm oils), and improving and selecting starter microorganisms used in winemaking (collaboration with the ISVV and the BioLaffort company).
4.1.1 Oil Palm lipid synthesis
The largest source of vegetable oil 1 is the fruit mesocarp of the oil palm Elaeis guineensis, a remarkable tissue that can accumulate up to 90% oil, the highest level observed in the plant kingdom. The market share of oil palm is expected to increase in order to meet increased demand for vegetable oil, predicted to double by 2030 27, be it as food or as a source of biofuels in Africa. A significant proportion of palm oil is produced on small estates that do not have access to efficient milling facilities, and run a great risk of spoilage through oil acidification. Improving palm oil quality through genetics and selection will result in economic gains 37 by addressing several targets such as improvement of oil yield, tuning of oil quality through the rate of unsaturated fatty acids or impairment of degradation processes. Furthermore, as genome biodiversity resides mostly in Africa, oil from African oil palms can vary greatly in fatty acid composition according to cultivar genetic differences and to weather conditions, and the precise mechanisms regulating this variability are not yet understood.
A growing body of molecular resources for studying oil palm fruit are making it possible to study and improve the quality and quantity of oil produced by oil palms. In particular, these oils can vary greatly in fatty acid composition, and while the precise mechanisms regulating this variability are not completely understood, establishing a link between oil palm genotype and phenotype appears increasingly feasible. Pleiade will work with the CNRS/UB UMR 5200 (LBM), a laboratory with an established reputation in studying fatty acid metabolism in E. guineensis, to improve understanding of the links between genetic diversity and oil production, and participate in developing applications.
4.1.2 Engineering pico-algae
Docosahexaenoic acid (DHA) is an essential nutriment for human brain tissue and can only be obtained from marine or riverine fish that live on phytoplankton and zooplankton, since human neurons lack the delta desaturase required for de novo synthesis of DHA. 34 Unfortunately, fishing is become less and less a sustainable resource. Since phytoplankton and zooplankton are the ultimate source of DHA consumed, there is considerable interest in obtaining DHA directly rather than through the intermediary of fish. A very promising approach is through the bio-engineering of pico-algae.
In order to produce the long-chain polyunsaturated fatty acids (LC-PUFA) needed for human nutrition, it is necessary to precisely engineer the desaturases that produce them. Desaturases are enzymes responsible for the introduction of double bonds into fatty acids. Desaturases are specific in recognizing their substrates and in placing the double bond in the proper place. The desaturases that produce the LC-PUFA necessary for human nutrition are present only in some species.
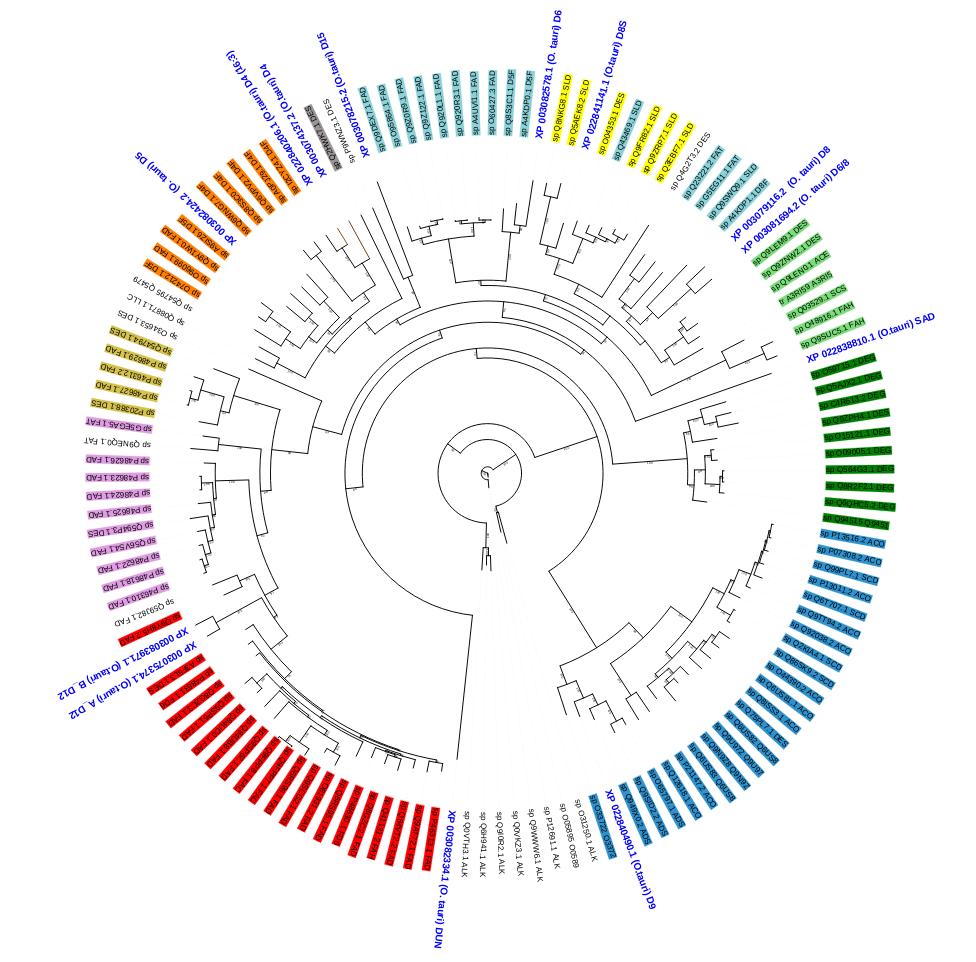
Our goal is to design methods to predict the substrate and region specificities for desaturases in algal species, particularly Ostreococcus tauri, the smallest photosynthetic eukaryote that can be cultivated. Thirteen desaturases are known in O. tauri and can be placed in the phylogeny of the desaturase family (figure 5). The biochemical and structural characterization of these enzymes is as yet very incomplete. This work requires close collaboration between biologists and computer scientists.
4.2 Molecular based systematics and taxonomy
Defining and recognizing myriads of species in biosphere has taken phenomenal energy over the past centuries and remains a major goal of Natural History. It is an iconic paradigm in pattern recognition (clustering has coevolved with numerical taxonomy many decades ago). Developments in evolution and molecular biology, as well as in data analysis, have over the past decades enabled a profound revolution, where species can be delimited and recognized by data analysis of sequences. We aim at proposing new tools, in the framework of E-science, which make possible () better exploration of the diversity in a given clade, and () assignment of a place in these patterns for new, unknown organisms, using information provided by sets of sequences. This will require investment in data analysis, machine learning, and pattern recognition to deal with the volumes of data and their complexity.
One example of this project is about the diversity of trees in Amazonian forest, in collaboration with botanists in French Guiana. Protists (unicellular Eukaryots) are by far more diverse than plants, and far less known. Molecular exploration of Eukaryotes diversity is nowadays a standard in biodiversity studies. Data are available, through metagenomics, as an avalanche and make molecular diversity enter the domain of Big Data. Hence, an effort will be invested, in collaboration with other INRIA teams (GenScale, HiePACS) for porting to HPC algorithms of pattern recognition and machine learning, or distance geometry, for these tools to be available as well in metagenomics. This will be developed first on diatoms (unicellular algae) in collaboration with INRAE team at Thonon and University of Uppsala, on pathogens of tomato and grapewine, within an existing network, and on bacterial communities, in collaboration with University of Pau. For the latter, the studies will extend to correlations between molecular diversity and sets of traits and functions in the ecosystem.
4.3 Community ecology and population genetics
Community assembly models how species can assemble or diassemble to build stable or metastable communities. It has grown out of inventories of countable organisms. Using metagenomics one can produce molecular based inventories at rates never reached before. Most communities can be understood as pathways of carbon exchange, mostly in the form of sugar, between species. Even a plant cannot exist without carbon exchange with its rhizosphere. Two main routes for carbon exchange have been recognized: predation and parasitism. In predation, interactions–even if sometimes dramatic–may be loose and infrequent, whereas parasitism requires what Claude Combes has called intimate and sustainable interactions 28. About one decade ago, some works 39 have proposed a comprehensive framework to link the studies of biodiversity with community assembly. This is still incipient research, connecting community ecology and biogeography.
We aim at developping graph-based models of co-occurence between species from NGS inventories in metagenomics, i.e. recognition of patterns in community assembly, and as a further layer to study links, if any, between diversity at different scales and community assemblies, starting from current, but oversimplified theories, where species assemble from a regional pool either randomly, as in neutral models, or by environmental filtering, as in niche modeling. We propose to study community assembly as a multiscale process between nested pools, both in tree communities in Amazonia, and diatom communities in freshwaters. This will be a step towards community genomics, which adds an ecological flavour to metagenomics.
Convergence between the processes that shape genetic diversity and community diversity–drift, selection, mutation/speciation and migration–has been noted for decades and is now a paradigm, establishing a continuous scale between levels of diversity patterns, beyond classical approaches based on iconic levels like species and populations. We will aim at deciphering diversity pattern along these gradients, connecting population and community genetics. Therefore, some key points must be adressed on reliability of tools.
Next-generation sequencing technologies are now an essential tool in population and community genomics, either for making evolutionary inferences or for developing SNPs for population genotyping analyses. Two problems are highlighted in the literature related to the use of those technologies for population genomics: variable sequence coverage and higher sequencing error in comparison to the Sanger sequencing technology. Methods are developed to develop unbiased estimates of key parameters, especially integrating sequencing errors 36. An additional problem can be created when sequences are mapped on a reference sequence, either the sequenced species or an heterologous one, since paralogous genes are then considered to be the same physical position, creating a false signal of diversity 31. Several approaches were proposed to correct for paralogy, either by working directly on the sequences issued from mapped reads 31 or by filtering detected SNPs. Finally, an increasingly popular method (RADseq) is used to develop SNP markers, but it was shown that using RADseq data to estimate diversity directly biases estimates 20. Workflows to implement statistical methods that correct for diversity biases estimates now need an implementation for biologists.
5 Highlights of the year
5.1 Clémence Frioux
Clémence Frioux, Inria staff junior scientist with a doctorate in Computer Science and an undergraduate degree in Biology, joined Pleiade in 2020. With her arrival we have considerably widened the scope of our work. Clémence brings a particular interest in community-scale metabolic modeling (§3.3) and a methodological focus on optimization techniques employing Answer Set Programming.
5.2 ADT Gordon
This year marked the delivery of the ADT Gordon in collaboration with HiePACS, Tadaam, and Storm. In general terms ADT Gordon consolidated the Inria HPC stack and in particular for Pleiade implemented random projection methods for multi-dimensional scaling. The success of ADT Gordon also led to two new projects that started this year: ADT Diodon, which will develop SVD methods for very large dimensionality reduction problems, and a preparatory project for PRACE with Hawk, HLRS, Stuttgart.
5.3 Health Crisis
The year 2020 was marked by the Covid crisis and its impact on society. The scientific world was also greatly affected: Faculty members have seen their teaching load increase significantly; PhD students and post-docs have often had to deal with a worsening of their working conditions, as well as with reduced interactions with their supervisors and colleagues; and most scientific collaborations have been greatly affected, with many international activities cancelled or postponed sine die.
6 New software and platforms
6.1 New software
6.1.1 Metage2Metabo
- Keywords: Metabolic networks, Microbiota, Metagenomics, Workflow
- Scientific Description: Flexible pipeline for the metabolic screening of large scale microbial communities described by reference genomes or metagenome-assembled genomes. The pipeline comprises several main steps. (1) Automatic and parallel reconstruction of metabolic networks. (2) Computation of individual metabolic potentials (3) Computation of collective metabolic potential (4) Calculation of the cooperation potential described as the set of metabolites producible by species only in a cooperative context (5) Computation of minimal-sized communities sastifying a metabolic objective (6) Extraction of key species (essential and alternative symbionts) associated to a metabolic function
- Functional Description: Metabolic networks are graphs which nodes are compounds and edges are biochemical reactions. To study the metabolic capabilities of microbiota, Metage2Metabo uses multiprocessing to reconstruct metabolic networks at large-scale. The individual and collective metabolic capabilities (number of compounds producible) are computed and compared. From these comparisons, a set of compounds only producible by the community is created. These newly producible compounds are used to find minimal communities that can produce them. From these communities, the keytstone species in the production of these compounds are identified.
- News of the Year: (1) Improvements of the pipeline and its continuous integration (2) Release of production-ready versions (3) Development of m2m-analysis subpipeline
-
URL:
https://
github. com/ AuReMe/ metage2metabo - Publication: hal-02395024
- Contact: Clémence Frioux
- Participants: Clémence Frioux, Arnaud Belcour, Anne Siegel
6.1.2 MiSCoTo
- Name: Microbiota Screening and COmmunity Selection with TOpology
- Keywords: Metabolic networks, ASP - Answer Set Programming, Logic programming
- Scientific Description: MiSCoTo solves combinatorial problems using Answer Set Programming. It aims at minimizing either the number of selected species or both the number of selected species and the cost of the interaction between them, characterized by the number of metabolic exchanges. In the first case, the level of modeling is called lumped or mixed-bag, in the latter, it is compartmentalized.
- Functional Description: Metabolic networks are composed of biochemical reactions and gather the expected metabolic capabilities of species. For organisms that live in interaction altogether (microbiotas), complementarity between these networks can be exploited to predict cooperation events. This software takes as inputs metabolic networks for various species (host, symbionts of the microbiota), components of the growth medium and a metabolic objective (metabolites to be produced), and aims at selecting a minimal set of symbionts to ensure the metabolic objective can be achieved. The software can use two types of modelings: a simplified one and another that takes into account the cost of metabolic exchanges and aims at minimizing it.
- Release Contributions: Memory usage optimization. Fix issues with input file formats.
- News of the Year: (1) Optimization of memory usage. (2) Add the possibility to create an output file (json format). (3) Fix issue with input file formats. (4) Minor changes in code and continuous integration (5) Improvement of outputs
-
URL:
https://
github. com/ cfrioux/ miscoto - Publication: hal-01871600
- Contact: Clémence Frioux
- Participants: Clémence Frioux, Anne Siegel, Enora Fremy, Camille Trottier, Arnaud Belcour
6.1.3 MeneTools
- Name: Metabolic networks Topological tools
- Keywords: Metabolic networks, Graph, Topology, Bioinformatics, Systems Biology, ASP - Answer Set Programming
- Scientific Description: MeneTools are a set of tools for the exploration of the producibility potential in a metabolic network using the network expansion algorithm. The MeneTools can: - assess whether targets are producible starting from nutrients (Menecheck) - get all compounds that are producible starting from nutrients (Menescope) - get all reactions that are activable from nutrients (Meneacti) - get production paths of specific compounds (Menepath) - obtain compounds that if added to the nutrients, would ensure the producibility of targets (Menecof) - identify metabolic deadends, i.e. metabolites that act as reactants of reactions but never as products, or metabolites that act as products of reactions but never as reactants. This is a purely structural analysis. All MeneTools using modelling follow the producibility in metabolic networks as defined by the network expansion algorithm.
- Functional Description: MeneTools consists in four topological tool to analyze metabolic models in a graph-based perspective. Menecheck verifies the producibility of target compounds from available substrates (growth medium) of the metabolic network. Menescope gives the whole range of accessible compounds in the metabolic network starting from substrates. Menepath give the production paths of given compounds in the model. Menecof proposes compounds that need to be produced or added as substrate for ensuring the producibility of targets.
- News of the Year: (1) reorganising outputs, including providing file outputs (2) implementation of deadend research in metabolic networks (3) implementation of the search for activable reactions (4) new command line calls (5) use clyngor instead of pyasp (deprecated). (6) better implementation of logs
-
URL:
https://
github. com/ cfrioux/ MeneTools - Publications: hal-01819150, hal-02395024
- Contact: Clémence Frioux
- Participants: Clémence Frioux, Anne Siegel, Arnaud Belcour
6.1.4 Fluto
- Keywords: ASP - Answer Set Programming, Answer Set Programming, Metabolic networks, Flux Balance Analysis, Linear programming
- Scientific Description: Fluto performs metabolic network completion with respect to topological and linear reaction rate constraints based on the stoichiometry of metabolic reactions.
- Functional Description: Fluto relies on Answer Set Programming (ASP) and a hybrid modelling that associates to ASP a Linear Programming (LP) constraint propagator. Models satisfying the qualitative constraints of network expansion are tested for satisfiability of flux constraints with the LP propagator. Resulting answer sets permit the completion of a metabolic network that ensures the metabolic reaction of interest is activated according to both formalisms.
- News of the Year: Reorganisation of the code. Implementation of continuous integration. Addition of the Sagot & Acuna formalism in the software.
-
URL:
https://
github. com/ cfrioux/ fluto/ - Publications: hal-01936778, hal-01557347
- Contact: Clémence Frioux
- Participant: Sven Thiele
- Partners: Max Planck Institute Magdeburg, University of Potsdam
6.1.5 Biodiversiton
- Name: Biodiversiton
- Keywords: Biodiversity, Comparative metagenomics, Clustering, Dimensionality reduction, Masses of data
- Functional Description: Biodiversiton is a suite of tools for biodiversity composed by Rsyst, pairwise_dis, diagno_syst, and yapotu. The global project provides tutorials, datasets, and a readme for the whole suite.
-
URL:
https://
gitlab. inria. fr/ metabarcoding/ biodiversiton - Authors: Alain Franc, Jean-Marc Frigerio, Franck Salin
- Contact: Alain Franc
6.1.6 Rsyst
- Name: Rsyst
- Keywords: Biodiversity, Metagenomics, Clustering, Dimensionality reduction, Masses of data
- Functional Description: Contains the R-Syst databases, in sqlite format, as well as python programs for querying them through a python interface for the most common queries.
-
URL:
https://
gitlab. inria. fr/ metabarcoding/ rsyst - Authors: Jean-Marc Frigerio, Franck Salin, Alain Franc
- Contact: Alain Franc
- Partner: INRAE
6.1.7 Pairwise_dis
- Name: Pairwise_dis
- Keywords: Biodiversity, Metagenomics, Clustering, Dimensionality reduction, Masses of data
-
Functional Description:
Routines in C and MPI versions for computing pairwise distances between reads as edit distances:
disseq: a C program whch runs as stand-alone, and can compute distance matrices between a few thousands of reads
mpidisseq: a parallelized version of disseq with MPI, which can compute distance matrices up to 150 000 reads within one day on a National Computing Center (e.g. a hyperparallel machine like a Blue Gene Q) , this program scales perfectly with the number of cores.
-
URL:
https://
gitlab. inria. fr/ metabarcoding/ pairwise_dis - Authors: Jean-Marc Frigerio, Alain Franc, Franck Salin
- Contact: Alain Franc
- Partner: INRAE
6.1.8 pydiodon
- Name: Pydiodon
- Keywords: Dimensionality reduction, Data analysis
-
Functional Description:
Most dimension reduction methods inherited from Multivariate Data Analysis, and currently implemented as elements in statistical learning for handling very large datasets (meaning the dimension of spaces is the number of features), rely on a chain of pretreatments, a core with a SVD for low rank approximation of a given matrix, and a post-treatment for interpreting results. The costly part in computations is the SVD, which is in cubic complexity. Diodon is a list of functions and drivers which implement (i) pre-treatments, SVD and post-treatments on a large diversity of methods, (ii) random projection methods for running the SVD which permits to bypass the time limit in computing the SVD, and (iii) an implementation in C++ of the SVD with random projection at prescribed rank or precision, connected to MDS.
Pydiodon is a deliverable of the ADT Diodon (see https://gitlab.inria.fr/diodon) which will provide an API in python (pydiodon) and C++ (cppdiodon), the former developed by Pleiade with the SED, the latter developped by the SED with Hiepacs (connections with FMR).
- News of the Year: In 2020, ADT Diodon has started with a fresh version of diodon as a starting point: new project in inria gitlab, renamed
-
URL:
https://
gitlab. inria. fr/ diodon/ pydiodon - Contact: Alain Franc
- Participants: Alain Franc, Jean-Marc Frigerio, Franck Salin, Florent Pruvost
- Partner: INRAE
6.1.9 Yapotu
- Name: Yet Another Pipeline for OTU building
- Keywords: Metagenomics, Biodiversity, Dimensionality reduction, Masses of data
- Functional Description: The main functionalities are as follows: 1) building OTUs from a fasta file (swarm, vsearch, ..) or a distannce file (yapotu) for an environmenal sample 2) building a fasta file and a distance file per OTU 3) checking the consistency of the OTUs by displaying them as a graph (see OTU as a graph below) 4) displaying the shape of an OTU or of a set of OTUs by Multidimensional Scaling 5) implementing Hierachical Aggregative Clustering of an OTU or a set of OTUs with various aggregation methods
- News of the Year: Ugraded from an older version, fusion with declic now deprecated, new functionalities for working with massive data sets
-
URL:
https://
gitlab. inria. fr/ metabarcoding/ yapotu - Authors: Alain Franc, Jean-Marc Frigerio, Franck Salin
- Contact: Alain Franc
- Partner: INRAE
6.1.10 Alcyone
- Name: Alcyone instantiates bioinformatics environments from specifications committed to a Git repository
- Keywords: Docker, Orchestration, Bioinformatics, Microservices, Versioning
- Scientific Description: Alcyone conceives the user's computing environment as a microservices architecture, where each bioinformatics tool in the specification is a separate containerized Docker service. Alcyone builds a master container for the specified environment that is responsible for building, updating, deploying and stopping these containers, as well as recording and sharing the environment in a Git repository. The master container can be manipulated using a command-line interface.
- Functional Description: Alcyone defines a file structure for the specifying bioinformatics analysis environments, including tool choice, interoperability, and sources of raw data. These specifications are recorded in a Git repository. Alcyone compiles a specification into a master Docker container that deploys and orchestrates containers for each of the component tools. Alcyone can restore any version of an environment recorded in the Git repository.
- News of the Year: Alcyone is being re-engineered to work on hosted Kubernetes platforms
-
URL:
https://
team. inria. fr/ pleiade/ alcyone/ - Contact: David James Sherman
- Participants: Louise-Amelie Schmitt, David James Sherman
6.1.11 Mimoza
- Keywords: Systems Biology, Bioinformatics, Biotechnology
- Functional Description: Mimoza uses metabolic model generalization and cartographic paradigms to allow human experts to explore a metabolic model in a hierarchical manner. Mimoza generalizes genome-scale metabolic models, by factoring equivalent reactions and metabolites while preserving reaction consistency. The software creates an zoomable representation of a model submitted by the user in SBML format. The most general view represents the compartments of the model, the next view shows the visualization of generalized versions of reactions and metabolites in each compartment , and the most detailed view visualizes the initial model with the generalization-based layout (where similar metabolites and reactions are placed next to each other). The resulting map can be explored on-line, or downloaded in a COMBINE archive. The zoomable representation is implemented using the Leaflet JavaScript library for mobile-friendly interactive maps. Users can click on reactions and compounds to see the information about their annotations.
- News of the Year: Mimoza is now available as a Docker image, and can be integrated with other containerized services using Pleiade's Alcyone system.
-
URL:
http://
mimoza. bordeaux. inria. fr/ - Publications: hal-00925881, hal-00859437, hal-00906911
- Contact: David James Sherman
- Participants: Anna Zhukova, David James Sherman
6.1.12 magecal
- Keyword: Genomics
- Scientific Description: Magecal independently runs training and prediction steps for Augustus, Conrad, GeneID, GeneMark, and Snap. The results are cleaned and integrated into a common format. Jigsaw is trained and used for model reconciliation. Consistency constraints are applied to ensure that phase and intron structure are biologically plausible.
- Functional Description: Magecal predicts a set of protein coding genes in fungal genomic sequences, using different de novo prediction algorithms, and reconciling the predictions with the aid of comparative data. Magecal applies consistency constraints to guarantee that the predicted genes are biologically valid.
- Release Contributions: Dockerization and compatibility with Alcyone
-
URL:
https://
gitlab. inria. fr/ magecal/ magecal - Contact: David James Sherman
- Participants: Pascal Durrens, David James Sherman
6.1.13 Magus
- Keywords: Bioinformatics, Genomic sequence, Knowledge database
- Scientific Description: MAGUS can be used on small installations with a web server and a relational database on a single machine, or scaled out in clusters or elastic clouds using Apache Cassandra for NoSQL data storage and Apache Hadoop for Map-Reduce.
- Functional Description: The MAGUS genome annotation system integrates genome sequences and sequences features, in silico analyses, and views of external data resources into a familiar user interface requiring only a Web navigator. MAGUS implements annotation workflows and enforces curation standards to guarantee consistency and integrity. As a novel feature the system provides a workflow for simultaneous annotation of related genomes through the use of protein families identified by in silico analyses this has resulted in a three-fold increase in curation speed, compared to one-at-a-time curation of individual genes. This allows us to maintain standards of high-quality manual annotation while efficiently using the time of volunteer curators.
- News of the Year: Magus is available as a Docker image, and can be integrated with other containerized services using Pleiade's Alcyone system.
-
URL:
http://
magus. gforge. inria. fr - Publication: inria-00563533
- Contact: David James Sherman
- Participants: David James Sherman, Florian Lajus, Natalia Golenetskaya, Pascal Durrens, Xavier Calcas
- Partners: Université de Bordeaux, CNRS
6.1.14 family-3d
- Keywords: Biodiversity, Point cloud, 3D modeling
- Scientific Description: The method statistically selects a subset of pairwise distances between proteins in the family, constructs a weighted graph, and lays it out using an adaptation of the three-dimensional extension of the Kamada-Kawai force-directed layout.
-
Functional Description:
Family-3D lays out high-dimension protein family point clouds in 3D space. The resulting lower-dimension forms can be printed, so that they can be explored and compared manually. They can also be explored interactively or stereographically.
Comparison of the 3D forms reveals classes of structurally similar families, whose characteristic shapes correspond to different evolutionary scenarios. Some of these scenarios are: neofunctionalization, subfunctionalization, founder gene effect, ancestral family.
To facilitate curator training, Family-3D includes an interactive terminal containing a microcontroller, an RFID reader, and an LED ring. A set of shapes that fall in predetermined classes is printed, with a unique RFID tag in each shape. Trainees classify family shapes by manual inspection and submit their classes to the terminal, which evaluates the proposed class and provides visual feedback.
-
URL:
https://
gitlab. inria. fr/ pleiade/ family-3d - Contact: David James Sherman
- Participant: David James Sherman
6.1.15 Diagno-Syst
- Name: diagno-syst: a tool for accurate inventories in metabarcoding
- Keywords: Biodiversity, Clustering, Ecology
- Functional Description: Diagno-syst builds accurate inventories for biodiversity. It performs supervised clustering of reads obtained from a next-generation sequencing experiment, mapping onto an existing reference database, and assignment of taxonomic annotations.
- Publication: hal-01426764
- Contact: Alain Franc
- Participants: Alain Franc, Jean-Marc Frigerio, Philippe Chaumeil, Franck Salin
- Partner: INRAE
7 New results
7.1 Characterization of Molecular Biodiversity
In 2020, Pleiade continued the development and refinement of new methods for chacterizing molecular biodiversity. Two approaches are being pursued, each with a PhD student in their second year.
- The central focus of Mohammed Anwar Abouabdallah's PhD 15 is building OTUs from a pairwise distance matrix using Stochastic Block Models (SBM). Building OTUs is traditionally seen as a form of unsupervised clustering. This work is done in collaboration with the MIAT INRAE research unit in Toulouse and HiePACS. It represents a connection between metabarcoding and statistical modeling, a topic which deserves investigation and is expanding (Figure 6 from 38).
- A major goal of Pleiade is to develop a geometric view of biodiversity. The tool selected up to now is to associate a point cloud to a dataset (pairwise distances between sequences) and to study its shape. This approach has expanded and been developed in 2020 as a collaboration with HiePACS through the cosupervision of Romain Peressonni's PhD, which aims to provide new approaches and algorithms for computing distances between two point clouds.
7.2 Dimension Reduction
Metabarcoding is a series of technical procedures to build molecular based inventories from large datasets of amplicons. The underlying information needs to be compacted without losing its information content before it can be further processed with domain-specific tools. This links metabarcoding tools to dimension reduction techniques, which is an important topic in Pleiade. This has been implemented through a participation in following research projects:
- Contribution to and finalization of the ADT Gordon project in Inria BSO. The objective of this project (partners: Tadaam (coordinator), Storm, HiePACS, Pleiade) is to integrate SVD as a tool available in Chameleon, starPU and new Madeleine. The contribution of Pleiade is to bring in metabarcoding as a use case, and random projection as a method for scaling Multidimensional Scaling (which requires an SVD) in collaboration with HiePACS with a template implemented in Diodon. A MDS on a 106 x 106 matrix has been succesfully run at the end of ADT Gordon, on Occigen, in 900 seconds including I/O. The final report has been issued by Tadaam in December 2020.
- A consequence of this involvement is the submission in 2020 of a new ADT, called Diodon, for extending to a diversity of linear dimension reduction techniques what has been aquired in ADT Gordon for MDS, namely a significant progress in speed and memory management brought by random SVD, which can be integrated into a diversity of methods : PCA, CoA, etc.
- Pleiade is involved in the EU project EOSC-Pillar (§8.2.1), in a task for better connecting data to calculation, currently data in Inrae Dataverse system connected to tools running on a INRAE local server or on PlaFRIM, on a testbed on biodiversity assessment with metabarcoding. This task in done in collaboration with INRAE DipSO (Direction Science Ouverte) and the Inria HiePACS project-team.
7.3 Clavispora lusitaniae genome assembly
New Clavispora lusitaniae strains have been isolated in patients by our LMFP partner (Laboratoire de Microbiologie Fondamentale et Pathogénicité, UMR-CNRS 5234) as well as Candida auris strains, this pathogen species being phygenetically very close to Clavispora lusitaniae. To the contrary of C. lusitaniae which is sensitive to antifungal drugs, C. auris is multi-drug resistant and a threat for the treatment of nosocomial diseases. In order to investigate on the determinism of resistance/sensitivity to antifungal drugs we need a reference genome sequence of Clavispora lusitania as complete as possible. High throughput Nanopore sequencing was performed on DNA extracted from 5 strains at Genotoul and assembled with Oxford Nanopore's MEDAKA tool. The assemblies turned to aberrant in size and structure. We tested another tool, CANU, and obtained assemblies compatible with what was expected from experimental measures. Unfortunately, error correction functions of CANU create mutations in the sequences, giving rise to non-functional genes, whatever the parameters used. We propose to override the problem by producing hybrid assemblies using other tools.
7.4 Genome Wide Association Study of oil production in palm tree
This work is a collaboration between Pleiade and the Vitapalm consortium funded by LEAP‑Agri, a joint Europe–Africa Research and Innovation initiative related to Food and Nutrition Security and Sustainable Agriculture. We tested and simulated several strategies for GWAS of oil production by the palm tree. We finally retained the reference panel / imputation panel strategy where the reference panel consists in 60 genomes sequenced at high coverage (15X) giving rise to a map of single nucleotide polymorphisms (SNP) which represent biodiversity within african palm trees ; other genomes (>300) will be sequenced at low coverage (0.5X) and will form the imputation panel where association between SNP and phenotype will be searched. Pleiade will be in charge of sequence treatment and SNP calling. Meanwhile other members of the Vitapalm consortium are making phenotypical measures on oil and vitamin compositions.
7.5 Multi-omic analysis of a cheese-derived bacterial community
Understanding and controlling the interactions within bacterial communities has applications in multiple industrial domains, among which the food processing industry. The TANGO project, conducted by the INRAE department STLO (Rennes) aimed at following a controlled bacterial community during the process of cheese production. The project also involved studying the impact of changes in production processes on organoleptic properties of the cheese. Multi-omic data was generated all along the experiment, enabling the monitoring of gene expression in bacteria, but also the metabolite production in the cheese. Pleiade was involved in the bioinformatic analysis of the project through the internship of Maxime Lecomte, in collaboration with Hélène Falentin (INRAE STLO Rennes) and Simon Labarthe (external collaborator of the team).
During his internship Maxime Lecomte contributed to building metabolic models for the members of the TANGO bacterial community, and performed the first simulations of the community metabolism over time.
The data generated by TANGO is a a great resource to study the evolution of a community over time. Such data now constitutes a basis for the PhD project of Maxime Lecomte. It aims at developping hybrid - logical and quantitative - modelling methods of the microbial metabolism in ecosystems. Calibrating such methods on a controlled and small scale ecosystem will be valuable for furture scaled up predictions.
7.6 Metabolic analyses of marine algae
Brown algae, especially the species Ectocarpus siliculosus, are important models for deciphering the complex interactions within marine holobionts, with the goal of studying their metabolism together with the metabolism of the bacteria that inhabit their direct environment. Because performing wet lab experiments on such systems is technocally challenging, there is need for bioinformatic predictive methods for assessing the putative roles and interdependences between species. In parallel, addressing the difficulties brought by the study of these organisms is also a means to enhance and calibrate the tools devlopped in the team. Hence a fruitful collaboration for the past years has been developed with scientists from the Roscoff Biological station.
In 2020, the genome of Ectocarpu subulatus was published 11, constituting a valuable resource for future studies that involve this stress-tolerant alga. In addition, we published a direct application of our methods for minimal community selection 10. In this work, we illustrate how predictions performed with MiSCoTo, a tool developped in the team, can meaningfully suggest metabolic dependencies between an alga and associated bacteria, and can help build controlled communities for laboratory cultures. In 2020, we also summarized in a review paper how the use of combinatorial optimisation problems such as the one solved in MiSCoTo can be applied to elucidate the physiology of brown algae 13.
7.7 Large-scale analyses of microbiomes diversity
With the decreasing cost of shotgun metagenomic experiments, the DNA sequences of more and more microbiotas become available, constituting an invaluable resource for deciphering their organisation. The growing resources of available metagenomes for a variety of ecosystems make it possible to study the distribution of bacteria and fungi environmental or host-associated microbiomes. We performed such work in 7 in which more than 13,000 metagenomes from 25 ecosystems were compiled. We demonstrated the differences in bacteria-to-fungi relative abundance ratio between environmental and host-associated microbiotas. We were able to distinguish habitats based on their composition in bacteria and fungi, highlighting differences between environmental habitats, external host and human influenced habitats, and anaerobic habitats like the gut.
In 14, we discussed the different possibilities for assessing the functions of an ecosystem starting from sequences. We evaluated the applicability and pitfalls of metabolic modelling in the context of metagenomes.
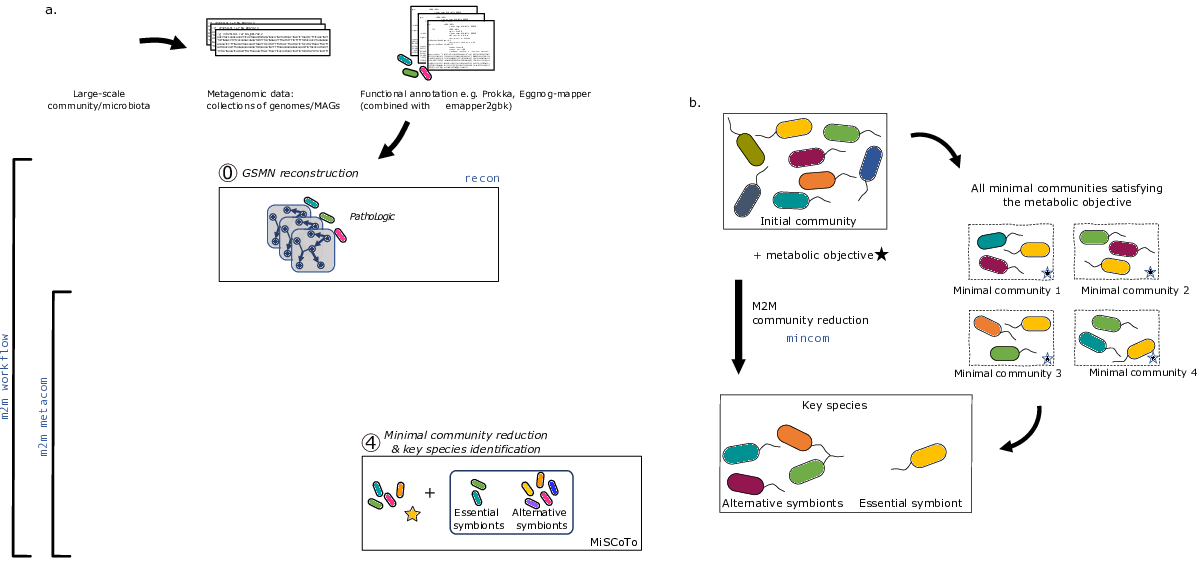
By assembling and binning the sequencing reads produced in metagenomics, it is possible to obtain metagenome-assembled genomes (MAGs) that can be assigned to taxonomic clades. These MAGs can be used to build predictions of the metabolism of the associated species, and in this way used as a proxy to understand the physiology of the underlying community. Metage2Metabo (M2M), a software system designed in Pleiade, aims at analysing the metabolic complementarity within a microbiota or a large collection of reference genomes, and to identify key species among them. Key species are members of the ecosystem that appear in some (alternative symbionts) or in all (essential symbionts) minimal communities associated to a function (see Figure 7 b.). M2M is a flexible pipeline that automatically performs metabolic network reconstruction from annotated genomes or MAGs, and analyses the resulting networks to capture the metabolic potential of associated species, both individually and as a community (see Figure 7 a.). We demonstrated the applicability of M2M using large-scale collections of genomes and MAGs, promoting the use of such a screening workflow to screen the metabolism of metagenomes 9. We presented M2M as a poster in the JOBIM conference (French conference gathering scientists of bioinformatics-related fields) in June 2020 18
In addition, we started the ADT MetagenoPic project, aiming at building a platform suitable for the analysis of raw metagenomic data, and bridging the resulting treated data to our existing methods for metabolic screening of communities (M2M).
8 Partnerships and cooperations
8.1 International initiatives
8.1.1 Participation in other international programs
Vitapalm – Food and nutrition security and sustainable agriculture in Africa
Pleiade participates in the Vitapalm program financed by LEAP-Agri2, the joint Europe Africa Research and Innovation (R&I) initiative related to Food and Nutrition Security and Sustainable Agriculture. Vitapalm uses genomics and selection to improve the nutritional quality and the stability of palm oil produced by Africa smallholdings for local consumption. Project partners are from Cameroon, France, Germany, and Ghana.
Simulation of metacommunities
In collaboration with the Pasteur Institute in Cayenne and the INRAE MIA Research Team in Toulouse, Pleiade is developing a stochastic model for simulation of metacommunities, in the framework of patch occupancy models. The objective is a better understanding of zoonose propagation, namely rabies through bat hosts in connection with disturbances of pristine forests in French Guiana, which have an impact on the exposure of human populations to wildlife that act as reservoirs of zoonoses.
CEBA – Center for the study of biodiversity in Amazonia
The Laboratoire of excellence CEBA promotes innovation in research on tropical biodiversity. It brings together a network of internationally-recognized French research teams, contributes to university education, and encourages scientific collaboration with South American countries. Pleiade participates in three current international projects funded by CEBA:
- MicroBIOMES: Microbial Biodiversities. 2017-19.
- Neutrophyl: Inferring the drivers of Neotropical diversification. 2017-19.
- Phyloguianas: Biogeography and pace of diversification in the Guiana Shield. 2015-present
Pleiade is involved with BioGeCo as partner of Institut Pasteur de Guyane at Cayenne for developing the domain of so-called Ecoviromics for some zoonoses in French Guiana. The spine of this collaboration is co-supervizing of a PhD student at IPG in cayenne, in bioinformatics and statistical ecology to decipher the respective roles of host phylogeny and environmetal variables in the virome of different hosts (bats, rodents, birds).
8.2 European initiatives
8.2.1 FP7 & H2020 Projects
PRACE 6th Implementation Phase Project
PRACE, the Partnership for Advanced Computing, is the permanent pan-European High Performance Computing service. High-performance Systems at Tier-0 are deployed by Germany, France, Italy, Spain and Switzerland, providing researchers with more than 17 billion core hours of compute time. PRACE-6IP assists the development of PRACE 2; strengthens the internationally recognised PRACE brand; provides advanced training; defines strategies and best practices for Exascale computing, works on forward-looking software solutions; coordinates and enhances the operation of the multi-tier HPC systems and services; and supports users in exploiting massively parallel systems and novel architectures.
EOSC-Pillar
Coordination and Harmonisation of National and Thematic Initiatives to support EOSC. This is a follow up of our former participation in EOSC-Pilot. In collaboration with HiePACS, Pleiade is involved in task 7.4, for bringing use cases in metabarcoding as testbeds for circulation of codes between different infrastructures, including PlaFRIM.
8.2.2 Collaborations in European programs, except FP7 and H2020
COST Action DNAqua.net
Pleiade is responsible for the WG “Data Analysis and storage” in this action. As such, in 2019 we organized with CNR Verbana (Italy) two European wide workshops: one in Lyon in February 2019, and one in Limassol (Cyprus) in October 2019. As a follow up of these workshops, Pleiade and BioGeCo became responsible for taking in charge data analysis of OTU picking in two European wide projects:
- a benchmark for different tools for OTU picking, with datasets from different European teams,
- a comparison between different organisms (metabarcoding inventories) for assessing the quality of the water of Danube river, in collaboration with raparian countries.
8.2.3 Collaborations with major European organizations
8.3 National initiatives
8.3.1 Agence Française pour la Biodiversité
The AFB is a public law agency of the French Ministry of Ecology that supports public policy in the domains of knowledge, preservation, management, and restoration of biodiversity in terrestrial, aquatic, and marine environments. Pleiade is a partner in two AFB projects developed with the former ONEMA: one funded by ONEMA, the second by labex COTE, where BioGeCo/Pleiade is responsible for data analysis, with implementaton of the tools recently developed for scaling MDS. Calculations have been made on CURTA at MCIA and PlaFRIM at INRIA.
8.4 Regional initiatives
Malabar
This is a project funded by labex COTE (University of Bordeaux) as a collaboration between EPOC (Talence), IFREMER (Arcachon), and ETI (chair of the Labex). The guideline of the project is to build models in statistical ecology on a series of molecular based invetories (300 samples) from occurence matrices of OTUs in samples, with environmental variables. The samples have been collected in 2018-2019, the sequences produced by BioGeCo in 2019, and data analysis will begin in 2020.
High-performance computing and metabarcoding
Pleiade is member of two projects, one funded by the Région Nouvelle Aquitaine and one funded as Inria ADT Gordon, connecting Chameleon, StartPU and NewMadeleine, where the use case of metabarcoding (questions, data sets) hase been selected to link these layers together. This will permit us to address unsupervised clustering of one million reads next year. These projects are in collaboration with the HiePACS, Tadaam, and Storm project-teams.
COTE – Continental to Coastal Ecosystems
The Labex cluster of excellence COTE (Continental To coastal Ecosystems: evolution, adaptability and governance) develops tools to understand and predict ecosystem responses to human-induced changes as well as methods of adaptative management and governance to ensure their sustainability. The LabEx includes nine laboratories of the University of Bordeaux and major national research institutes involved in research on terrestrial and aquatic ecosystems (INRAE, CNRS, and IFREMER).
9 Dissemination
9.1 Promoting scientific activities
9.1.1 Scientific events: organisation
Alain Franc of Pleiade is deeply involved in the organisation and leadership of COST programme DNAqua.net final General A1ssembly which takes the form of a (virtual) conference gathering about 1300 attendees with 250 submitted abstracts. Pleiade is involved as part of the core grop (Management Committee), Scentific Committee and co-head of working group on Data Analysis and storage.
Member of the editorial boards
Alain Franc is member of the editorial board of BMC Evolutionary Biology.
Pascal Durrens is a member of the editorial board of the journal ISRN Computational Biology.
Reviewer - reviewing activities
Clémence Frioux reviewed articles for the following journals: Bioinformatics, BioSystems, BMC Bioinformatics and Scientific Reports.
9.1.2 Invited talks
- Gap-filling metabolism in systems biology and microbial systems ecology - Clémence Frioux - BioNum Seminar - LaBRI Bordeaux - February 2020
9.1.3 Scientific expertise
David Sherman is a member of the Scientific Advisory Board of Enlightware GmbH, Zürich.
9.1.4 Research administration
Alain Franc has been appointed “chargé de mission calcul” at INRAE by the INRAE Delegate for Digital Transition. As such, his mission is to propose animations and solutions for the development of scientific computing at INRAE, whatever the Research Department.
David Sherman is president of the Commission for Technology Development (CDT) of the Inria Bordeaux Sud-Ouest research center. The CDT has two roles. First, it evaluates funding requests for Technology Development and Technology Transfer projects, which typically involve hiring technical staff. Second, the CDT is responsible for validating and overseeing contract engineers hired by Inria project-teams.
David Sherman is a member of the steering committee of the Region Nouvelle Aquitaine's regional research network “Biodiversity and Ecosystemic Services” (BIOSENA). BIOSENA unites academic and socio-professional actors who share the goal of contributing to knowledge and preservation of biodiversity, as wel as the improvement of services to ecosystems through research, knowledge dissemination, promoting scientific culture and transferring skills through research actions. BIOSENA implements the recommendations of Ecobiose.
9.2 Teaching - Supervision - Juries
9.2.1 Teaching
Clémence Frioux
- Master – ENSTBB Bordeaux INP - Bioinformatics
- Master – Master Bioinformatique Université de Bordeaux - Projet de Programmation
9.2.2 Supervision
- Internship Ariane Badoual (Master 2 Bioinformatique et Génomique Université de Rennes 1) - January to July 2020 - supervised by Clémence Frioux
- Internship Maxime Lecomte (Master 2 Bioinformatique Université de Bordeaux) - February to August 2020 - cosupervised by Clémence Frioux and Simon Labarthe
- PhD Maxime Lecomte - from November 2020 - cosupervised by Hélène Falentin, David Sherman and Clémence Frioux
9.2.3 Doctoral Advisory Committees
David Sherman was a member of the doctoral advisory committees of:
- Andony Arrieula
- Charlotte Herzog
9.3 Popularization
Unusually, Pleiade did not organize popularization activities during the 2020 calendar year, because of the Covis-19 crisis.
10 Scientific production
10.1 Major publications
- 1 articleA Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum.Nature Communications52014, 4044
- 2 articleModeling acclimatization by hybrid systems: Condition changes alter biological system behavior modelsBioSystems121June 2014, 43-53
- 3 article Metage2Metabo, microbiota-scale metabolic complementarity for the identification of key species eLife 9 December 2020
- 4 incollectionWhy We Need Sustainable Networks Bridging Countries, Disciplines, Cultures and Generations for Aquatic Biomonitoring 2.0: A Perspective Derived From the DNAqua-Net COST ActionNext Generation Biomonitoring: Part 158Elsevier2018, 63-99
- 5 articleExact or approximate inference in graphical models: why the choice is dictated by the treewidth, and how variable elimination can be exploitedAustralian and New Zealand Journal of Statistics612to appearJune 2019, 89-133
- 6 articleGenolevures: protein families and synteny among complete hemiascomycetous yeast proteomes and genomes.Nucleic Acids Research372009, D550-D554
10.2 Publications of the year
International journals
Conferences without proceedings
Scientific book chapters
Other scientific publications
10.3 Cited publications
- 19 articleSIGPLAN NoticesGenerating Embedded Software from Hierarchical Hybrid Models3872003, 171--82
- 20 articleRADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype samplingMol. Ecol.22112013, 3179--90
- 21 articleReusing and composing models of cell fate regulation of human bone precursor cellsBioSystems1081-3April 2012, 63-72
- 22 articleImplementing biological hybrid systems: Allowing composition and avoiding stiffnessApplied Mathematics and Computation223August 2013, 167--79
- 23 inproceedingsReconciling competing models: a case study of wine fermentation kineticsAlgebraic and Numeric Biology 20106479Research Institute for Symbolic Computation, Johannes Kepler University of LinzHagenberg, AustriaSpringerJuly 2010, 68--83
- 24 articleThe Euclidean Distance Matrix Completion ProblemSIAM J. Matrix Anal. App.1621995, 646-654
- 25 techreportA geometric view of Biodiversity: scaling to metagenomicsRR-9144https://arxiv.org/abs/1803.02272INRIA ; INRAJanuary 2018, 1-16
- 26 articleExact Matrix Completion via Convex OptimizationFound. Comput. Math.92009, 717-772
- 27 articleReplacing fossil oil with fresh oil - with what and for what?Eur J Lipid Sci Technol11372011, 812-831
- 28 book Parasitism: The Ecology and Evolution of Intimate Interactions University of Chicago Press 2001
- 29 articleStructural analysis of expanding metabolic networks.Genome informatics. International Conference on Genome Informatics1512004, 35--45
- 30 articleScalable and exhaustive screening of metabolic functions carried out by microbial consortiaBioinformatics34172018, i934--i943
- 31 article Reference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap PLoS Genetic 9 4 e1003457 2013
- 32 article Clingo = ASP + Control: Preliminary Report CoRR abs/1405.3694 2014
- 33 inproceedingsAdvances in gringo Series 3LPNMR6645Lecture Notes in Computer ScienceSpringer2011, 345---351
- 34 articleDocosahexaenoic acid: one molecule diverse functionsCrit Rev Biotechnol.375Aug 2017, 579-597URL: http://dx.doi.org/10.1080/07388551.2016.1207153
- 35 articleEuclidean Distance Geometry and ApplicationsSIAM review56(1)2014, 3-69
- 36 articleEstimation of Nucleotide Diversity, Disequilibrium Coefficients, and Mutation Rates from High-Coverage Genome-Sequencing ProjectsMol. Biol. Evol.25112008, 2409--19
- 37 articleImproving palm oil quality through identification and mapping of the lipase gene causing oil deteriorationNat Commun42013, 2160URL: http://dx.doi.org/10.1038/ncomms3160
- 38 articleExact or approximate inference in graphical models: why the choice is dictated by the treewidth, and how variable elimination can be exploitedAustralian and New Zealand Journal of Statistics612June 2019, 89-133
- 39 articleA comprehensive framework for global patterns in biodiversityEcology Letters712004, 1--15URL: http://dx.doi.org/10.1046/j.1461-0248.2003.00554.x
- 40 article R-Syst::diatom: an open-access and curated barcode database for diatoms and freshwater monitoring Database - The journal of Biological Databases and Curation 2016 February 2016
- 41 articleA unifying review of linear Gaussian ModelsNeural Computation1121999, 305--45
- 42 articleThink globally, fit locally: unsupervised learning of low dimensional manifoldsJournal of Machine Learning Research42003, 119--55
- 43 book On Growth and Form Cambridge University Press 1917
- 44 book Geometric structure of high-dimensional data and dimensionality reduction Springer & Higher Education Press 2012