
Keywords
Computer Science and Digital Science
- A2.1.1. Semantics of programming languages
- A2.1.5. Constraint programming
- A2.1.10. Domain-specific languages
- A2.2.1. Static analysis
- A2.3.2. Cyber-physical systems
- A2.4. Formal method for verification, reliability, certification
- A2.4.1. Analysis
- A2.4.2. Model-checking
- A2.4.3. Proofs
- A3.4.2. Unsupervised learning
- A3.4.4. Optimization and learning
- A6.1.1. Continuous Modeling (PDE, ODE)
- A6.1.2. Stochastic Modeling
- A6.1.3. Discrete Modeling (multi-agent, people centered)
- A6.1.4. Multiscale modeling
- A6.2.4. Statistical methods
- A6.2.6. Optimization
- A6.3.1. Inverse problems
- A6.3.4. Model reduction
- A7.2. Logic in Computer Science
- A8.1. Discrete mathematics, combinatorics
- A8.2. Optimization
- A8.7. Graph theory
- A9.7. AI algorithmics
Other Research Topics and Application Domains
- B1. Life sciences
- B1.1.2. Molecular and cellular biology
- B1.1.7. Bioinformatics
- B1.1.8. Mathematical biology
- B1.1.10. Systems and synthetic biology
- B2.2.3. Cancer
- B2.2.6. Neurodegenerative diseases
- B2.4.1. Pharmaco kinetics and dynamics
- B9. Society and Knowledge
1 Team members, visitors, external collaborators
Research Scientists
- François Fages [Team leader, Inria, Senior Researcher, HDR]
- Aurelien Naldi [Inria, Starting Research Position]
- Sylvain Soliman [Inria, Researcher, HDR]
Faculty Member
- Anna Niarakis [Univ d'Evry Val d'Essonne, Associate Professor, HDR]
PhD Students
- Sahar Aghakhani [Univ d'Evry Val d'Essonne]
- Eleonore Bellot [Inria, until Mar 2021]
- Marine Collery [IBM France]
- Elea Greugny [Johnson & Johnson, CIFRE]
- Jeremy Grignard [Servier]
- Julien Martinelli [INSERM]
Technical Staff
- Mathieu Hemery [Inria, Engineer]
Interns and Apprentices
- Martin Boutroux [Inria, until Apr 2021]
- Solene Mansour [Université Clermont Auvergne, from Jun 2021 until Jul 2021]
Administrative Assistant
- Natalia Alves [Inria]
External Collaborator
- Denis Thieffry [École Normale Supérieure de Paris, HDR]
2 Overall objectives
This project aims at developing formal methods for understanding the cell machinery and establishing computational paradigms in cell biology. It is based on the vision of
cells as machines,
chemical reaction networks as programs,
and on the use of concepts from computer science to master the complexity of cell biochemical processes1.
We contribute to the development of a computational theory of chemical reaction networks (CRNs), by addressing fundamental research issues in computer science on the concepts of analog computation and analog computational complexity in biochemistry, and on the interplay between structure and dynamics in CRNs.
Since 2002, we develop a software platform, called the Biochemical Abstract Machine (BIOCHAM), for modeling, analyzing and now synthesizing CRNs, with some unique algorithmic contributions. The reaction rule-based language of BIOCHAM allows us to reason about CRNs at different levels of abstraction in the hierarchy of their stochastic, differential, Boolean and hybrid semantics. Various static analysis methods, most of them based on constraint solving or graph theory, provide useful information before going to simulations and dynamical analyses, for which quantitative temporal logic is used to formalize cell behaviors with imprecise data, and to constrain model building.
A tight integration between dry lab and wet lab efforts is also essential for the success of the project. This is achieved through collaborations with biologists and experimentalists, including partners from the pharmaceutical industry, on concrete biological and biomedical questions.
Because of both the importance of constraint solving and optimization techniques in our approach, and the need of rapid prototyping for our software developments, we keep some research and teaching activity in constraint logic programming, as a general paradigm for computing with partial information systems, and solving practical NP-hard problems.
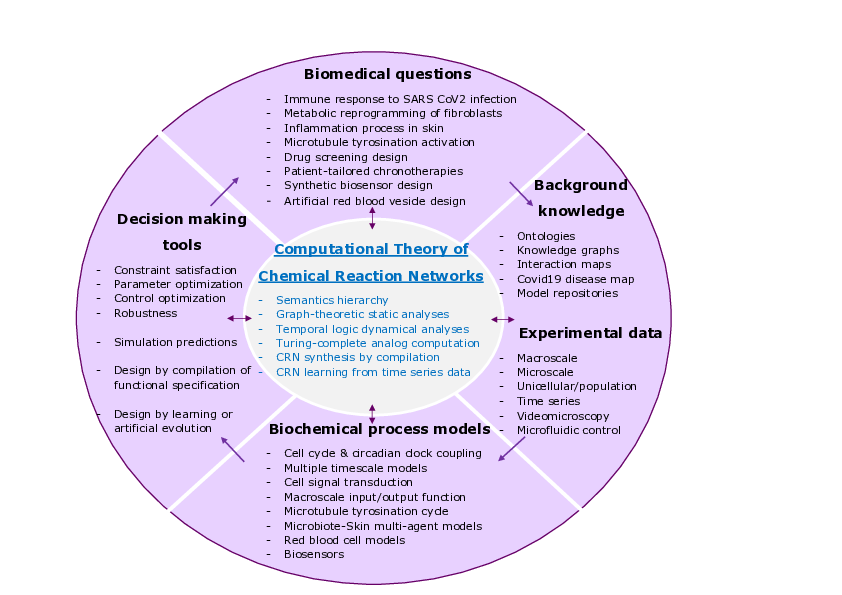
Wheel of biomedical questions, background knowledge and experimental data, biological process models and decision making tools, with the computational theory of chemical reaction networks at the center.
3 Research program
3.1 Chemical Reaction Network (CRN) Theory
Originally, Feinberg's CRN theory, and Thomas's influence networks analyses, were created to provide sufficient and/or necessary structural conditions for the existence of multiple steady states and oscillations in complex gene networks. Those conditions can be verified by static analyzers without knowing kinetic parameter values, nor making any simulation.
In this approach, most of our work consists in analyzing the interplay between the structure (Petri net invariants, influence graph, reductions by subgraph epimorphisms) and the dynamics (Boolean, CTMC, ODE, time scale separations) of CRNs in their different interpretations2. In particular, our study of the influence graphs of reaction systems lead to the non-trivial generalization of Thomas' conditions of multi-stationarity and Soulé's proof for influence systems, to reaction systems 34.
Furthermore, our original method to infer CRNs from ODEs 5 showed the generality of CRNs, and lead us to prove the Turing-completeness of continuous CRNs over a finite set of molecular species, showing that any computable function over the real numbers (i.e. computable in arbitrary precision by a Turing machine) can be computed by a CRN over a finite set of molecular species6 (Best paper award CMSB 2017, Prix La Recherche 2019). This result opens a whole research avenue on CRN design by compilation of mathematical functions 1, with innovative applications in synthetic biology7.
3.2 Logical Paradigm for Systems Biology
Our group was among the first ones in 2002 to apply model-checking methods to systems biology in order to reason on large molecular interaction networks, such as Kohn's map of the mammalian cell cycle (800 reactions over 500 molecules) 8. The logical paradigm for systems biology that we have subsequently developed for quantitative models can be summarized by the following identifications :
biological model = transition system
dynamical behavior specification = temporal logic formula
model validation = model-checking
model reduction = sub-model-checking, s.t.
model prediction = formula enumeration, s.t.
static experiment design = symbolic model-checking, state s.t.
model synthesis = constraint solving
dynamic experiment design = constraint solving
In particular, the definition of a continuous satisfaction degree for first-order temporal logic formulae with constraints over the reals, was the key to generalize this approach to quantitative models, opening up the field of model-checking to model optimization 910
This line of research continues with the development of temporal logic constraint patterns with efficient solvers, and their experimentation for fast model building, in partnership with biologists to answer concrete questions in the biomedical domain and the pharmaceutical industry11121314.
3.3 Constraint Solving and Optimization
Constraint solving and optimization algorithms are important in our research. On the one hand, static analyses of CRNs often involve solving hard combinatorial optimization problems, for which we have shown that constraint logic programming techniques are particularly successful, often beating dedicated algorithms on real-size instances from model repositories1516.
On the other hand, parameter search problems involve solving hard continuous optimization problems, for which evolutionary algorithms, and especially the covariance matrix evolution strategy (CMA-ES) 17 have shown to provide best results in our context. Constraint-based models and efficient constraint solvers are thus instrumental in our approach for building challenging quantitative models, gaining model-based insights, revisiting admitted assumptions, and contributing to biological knowledge.
4 Application domains
4.1 Preamble
Our collaborative work on biological applications is expected to serve as a basis for groundbreaking advances in cell functioning understanding, cell monitoring and control, and novel therapy design and optimization. Our collaborations with biologists are focused on concrete biological questions, and on the building of predictive computational models of biological systems to answer them. Furthermore, one important application of our research is the development and distribution of a modeling software for computational systems biology and synthetic biology.
4.2 Modeling software for systems and synthetic biology at the cellular level
Since 2002, we develop an open-source software environment for modeling and analyzing biochemical reaction systems. This software, called the Biochemical Abstract Machine (BIOCHAM), is compatible with SBML for importing and exporting models from repositories such as BioModels. It can perform a variety of static analyses, specify behaviors in Boolean or quantitative temporal logics, search parameter values satisfying temporal constraints, and make various simulations. While the primary reason of this development effort is to be able to implement our ideas and experiment them quickly on a large scale, using rapid prototyping techniques based on constraint logic programming, BIOCHAM is distributed and used by other groups worldwide, for building CRN models, for comparing CRN analysis/synthesis techniques, and for teaching computational systems biology. A Jupyter BIOCHAM kernel has been created to make it possible to use BIOCHAM on our web server without any installation. We plan to continue developing BIOCHAM for these different purposes and improve the software quality and animation of the community of users with the recruitment of a research engineer.
Since 2018, the CaSQ software complements this effort by providing an interface to import large interaction maps written in SBML using the CellDesigner tool and translate them into Boolean influence models with various tools compatible with the SBML-qual standard.
Since 2020, we participate in the CoLoMoTo notebook platform, which provides an integrated collection of software tools for the analysis of qualitative models, including CaSQ. This platform encourages the reproducibility of analysis by combining Docker images (reproducible software environment) and Jupyter notebooks (reproducible and shareable workflows).
These two last efforts play a central role in the global Covid-19 Disease Maps project, see Section 6.2.
4.3 CRN design and implementation in non-living protocells
In collaboration with Franck Molina (prix de l'innovation CNRS 2020), Lab. Sys2Diag ALCEN-CNRS, Montpellier, and Jie-Hong Jiang, NTU, Taiwan, we aim at applying our original CRN design pipeline to the implementation of high-level functions with enzymatic reactions in DNA-free non-living vesicles. The targeted medical applications concern the design of biosensors for medical diagnosis 18, and the design of artificial red blood protocells (Inria action AEx GRAM).
Our approach is based on analog computation theory, and our capability to compile mathematical functions and controllers in DNA-free, RNA-free biochemical reactions with abstract proteins. The concrete realizations are experimented using microfluidic devices at Sys2Diag Lab. in order to precisely control both the size of the vesicles and the concentrations of the injected chemical compounds. Interestingly, it is worht noting that the choice of non-living chassis, in contrast to living cells widely used in synthetic biology, is particularly appealing for many innovative applications involving security considerations and compliance to EU regulation.
4.4 Cell signaling networks
The unique features of BIOCHAM have been used in several success stories in the analysis, or now synthesis, of cell signaling networks. This was for instance the case for the deciphering of the complex dynamics of G protein-coupled receptors, using original parameter optimization techniques with temporal logic constraints19.
More recently, using our CRN compilation pipeline of mathematical functions, we could generate synthetic analogs of the natural and ubiquitous MAPK cell signaling network, by compilation of similar sigmoid input/ouput functions 1.
4.5 Coupled models of the cell cycle and the circadian clock
Recent advances in cancer chronotherapies support the evidence that there exist important links between the cell cycle and the circadian clock genes. One purpose for modeling these links is to better understand how to efficiently target malignant cells depending on the phase of the day and patient characterictics. These questions have been at the heart of a series of collaborative projects with Franck Delaunay (CNRS Nice) and Francis Lévi (INSERM Hopital Paul Brousse, Villejuif) and now Annabelle Ballesta (INSERM, Institut Curie Paris), with active participation to ANR Hyclock, EU EraNet Sysbio C5Sys and FP6 TEMPO projects.
In the past, we developed a coupled model of the Cell Cycle, Circadian Clock, DNA Repair System, Irinotecan Metabolism and Exposure Control under Temporal Logic Constraints 20, and a bidirectional coupled model of the cell cycle and the circadian clock to explain unexpected observations in single-cell experiments in fibroblasts21.
This year, several new results were obtained in the Thesis of Julien Martinelli 29, see Section 8.3.
5 Social and environmental responsibility
5.1 Footprint of research activities
In synthetic biology, our approach based on analog computation target enzymatic reactions with proteins in artificial vesicles which are thus DNA-free and RNA-free.
5.2 Impact of research results
Our multidisciplinary research rooted in fundamental computer science aims at contributing to biology and medicine by going quite far in the applications.
6 Highlights of the year
6.1 Compiling Elementary Mathematical Functions into Finite Chemical Reaction Networks via a Polynomialization Algorithm for ODEs
In 1, we present a polynomialization algorithm of quadratic time complexity to transform a system of elementary differential equations to polynomial ordinary differential equations (PODE). This algorithm is used as a front-end transformation in the compilation pipeline of BIOCHAM, to compile any elementary mathematical function, either of time or of some input species, into a finite CRN (see Fig. 2). For instance, the CRN synthesized for the Hill sigmoid function of order 5 provides a synthetic analog of the ubiquitous MAPK signalling CRN.
This work has been presented at CMSB 2021 1, CASC 2021 15 and in an invited talk given at SIAM AG 2021. It is a follow-up of our previous results on the Turing-completeness of continuous CRN over a finite set of molecular species, and on the complexity of the PODE quadratization problem.
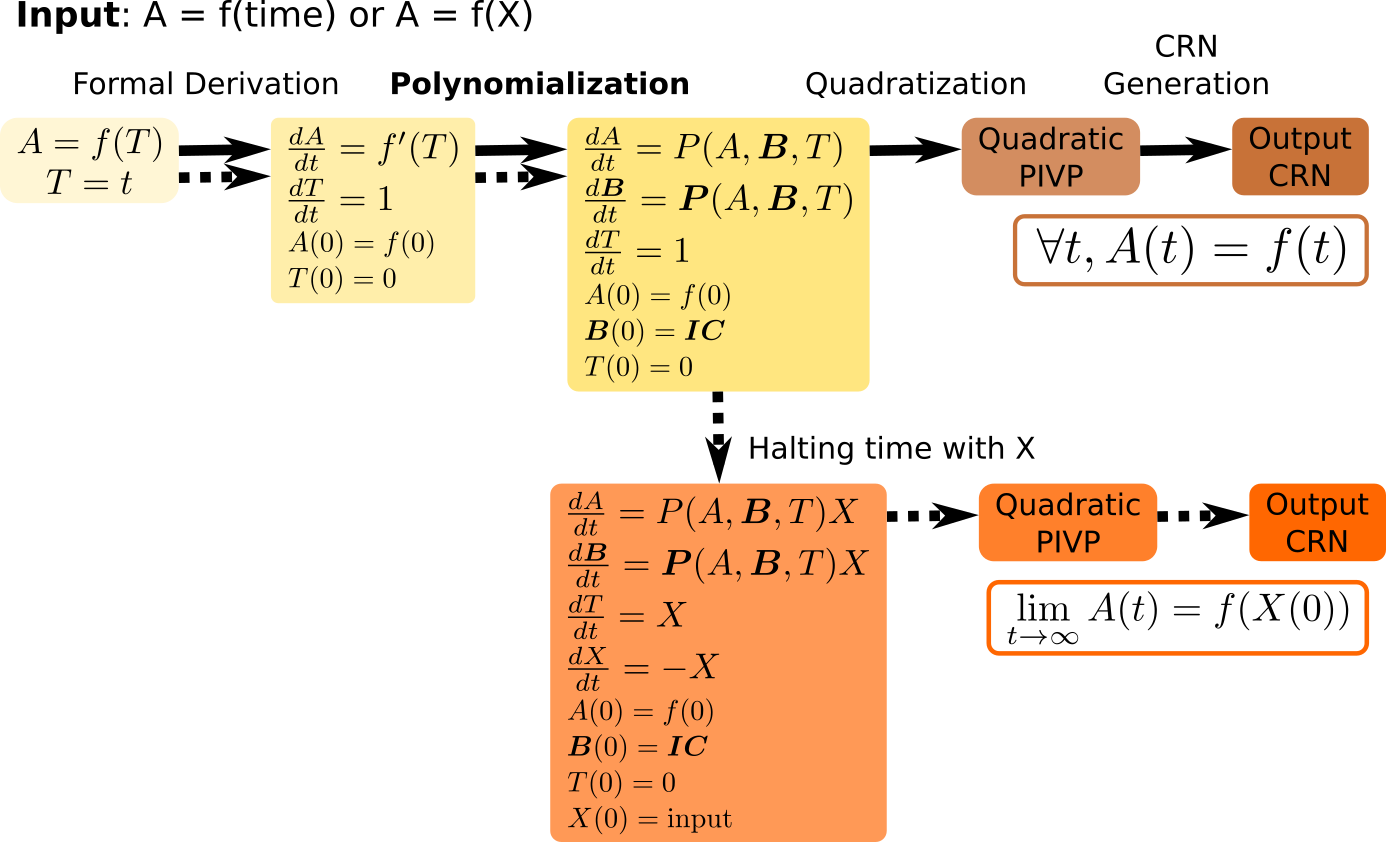
Chain of transformations to going from the symbolic representaiton of a function f(t) to an ODE of which f(t) is solution, to a polynomial ODE, to a quadratic ODE, and finally to a CRN implenting f(t) on one output molecular species.
6.2 Covid-19 disease map: a bioinformatics feat
The disease map of Covid-19 mentioned last year in highlight has been published this year in Mol. Sys. Biol.3.
7 New software and platforms
7.1 New software
7.1.1 BIOCHAM
-
Name:
The Biochemical Abstract Machine
-
Keywords:
Bioinformatics, Systems Biology, Computational biology
-
Functional Description:
The Biochemical Abstract Machine (BIOCHAM) is a software environment for modeling, analyzing and synthesizing biochemical reaction networks (CRNs) with respect to a formal specification of the observed or desired behavior of a biochemical system. BIOCHAM is compatible with the Systems Biology Markup Language (SBML) and contains some unique features about formal specifications in quantitative temporal logic, sensitivity and robustness analyses and parameter search in high dimension w.r.t. behavioral specifications, static analyses, and synthesis of CRNs.
-
Release Contributions:
– compilation pipeline of elementary mathematical functions in at most bi-molecular reaction networks over a finite set of abstract molecular species – improved heuristic quadratization algorithm to generate at most bi-molecular reactions – notebooks of Master classes in Bioinfomratics of molecular interaction networks – multiple improvements of the commands and documentation
- URL:
-
Contact:
Francois Fages
-
Participants:
Francois Fages, Mathieu Hemery, Sylvain Soliman
7.1.2 CaSQ
-
Name:
CellDesigner as SBML-Qual
-
Keywords:
SBML, Logical Framework, Knowledge representation
-
Functional Description:
CaSQ transforms a big knowledge map encoded as an SBGN-compliant SBML file in CellDesigner into an executable Logical Model in SBML-Qual
- URL:
-
Contact:
Sylvain Soliman
-
Participants:
Sylvain Soliman, Anna Niarakis, Aurélien Naldi
-
Partner:
Université d'Evry-Val d'Essonne
7.1.3 CoLoMoTo Notebook
-
Keywords:
Systems Biology, Computational biology
-
Functional Description:
Dynamical models are widely used to summarize our understanding of biological networks, to challenge its consistency, and to guide the generation of realistic hypothesis before experimental validation. Qualitative models in particular enable the study of the dynamical properties that do not depend on kinetic parameters, which can be difficult to obtain. This platform integrates a wide collection of complementary software tools, along with interoperability features and a consistent Programming interface based on the popular Python language.
The platform is distributed as a Docker image, ensuring the reproducibility of the software environment. The use of Jupyter Notebooks provides a good balance between ease of use and flexibility. These notebooks can be further used to share precise analysis workflows, ensuring the dissemination of fully reproducible research work using this platform.
- URL:
- Publication:
-
Contact:
Aurélien Naldi
-
Participants:
Sylvain Soliman, Aurélien Naldi
-
Partners:
LaBRI, Institut Curie, ENS Paris, LRI - Laboratoire de Recherche en Informatique
8 New results
8.1 Compiling Elementary Mathematical Functions into Finite Chemical Reaction Networks via a Polynomialization Algorithm for ODEs
Participants: François Fages, Mathieu Hemery, Sylvain Soliman.
The Turing completeness result for continuous chemical reaction networks (CRN) shows that any computable function over the real numbers can be computed by a CRN over a finite set of formal molecular species using at most bimolecular reactions with mass action law kinetics. The proof uses a previous result of Turing completeness for functions defined by polynomial ordinary differential equations (PODE), the dualrail encoding of real variables by the difference of concentration between two molecular species, and a back-end quadratization transformation to restrict to elementary reactions with at most two reactants. The presentation of a mathematical function as the solution of a PODE is however not always evident.In 1, 15, we present a polynomialization algorithm of quadratic time complexity to transform a system of elementary differential equations in PODE. This algorithm is used as a front-end transformation to compile any elementary mathematical function, either of time or of some input species, into a finite CRN (see Fig. 2). We illustrate the performance of our compiler on a benchmark of elementary functions relevant to CRN design problems in synthetic biology specified by mathematical functions. In particular, the abstract CRN obtained by compilation of the Hill function of order 5 is compared to the natural ubiquitous CRN structure of MAPK signalling networks.
8.2 Rule learning from time series data
Participants: Marine Collery, François Fages, Jeremy Grignard, Mathieu Hemery, Julien Martinelli, Sylvain Soliman.
With the automation of biological experiments and the increase of quality of single cell data that can now be obtained by phosphoproteomic and time lapse videomicroscopy, automating the building of mechanistic models from these data time series becomes conceivable and a necessity for many new applications. While learning numerical parameters to fit a given model structure to observed data is now a quite well understood subject, learning the structure of the model is a more challenging problem that previous attempts failed to solve without relying quite heavily on prior knowledge about that structure.
In a paper in preparation, we present an unsupervised statistical learning heuristic search algorithm to infer reaction rules with a time complexity for inferring one reaction in linear time in the number of observed transitions in the traces, and quadratic time in the number of observed molecular species.
In the thesis of Marine Collery at IBM France in the domain fraud detection from bank transaction traces, we have evaluated a somewhat similar rule inference algorithm in 17.
8.3 Model learning to identify systemic regulators of the peripheral circadian clock
Participants: François Fages, Julien Martinelli, Sylvain Soliman.
In 2, we present a model learning approach to identify systemic regulators of the peripheral circadian clock. Personalized medicine aims at providing patient-tailored therapeutics based on multi-type data towards improved treatment outcomes. Chronotherapy that consists in adapting drug administration to the patient's circadian rhythms may be improved by such approach. Recent clinical studies demonstrated large variability in patients' circadian coordination and optimal drug timing. Consequently, new eHealth platforms allow the monitoring of circadian biomarkers in individual patients through wearable technologies (rest-activity, body temperature), blood or salivary samples (melatonin, cortisol), and daily questionnaires (food intake, symptoms). A current clinical challenge involves designing a methodology predicting from circadian biomarkers the patient peripheral circadian clocks and associated optimal drug timing. The mammalian circadian timing system being largely conserved between mouse and humans yet with phase opposition, the study was developed using available mouse datasets.
We investigated at the molecular scale the influence of systemic regulators (e.g. temperature, hormones) on peripheral clocks, through a model learning approach involving systems biology models based on ordinary differential equations. To that end, we rely on prior knowledge encompassed in a circadian clock model that was also the subject of a publication in 9. The latter allowed us to derive an approximation for the action of systemic regulators on the expression of three core-clock genes: Bmal1, Per2 and Rev-Erb. These time profiles were then fitted with a population of models, based on linear regression. Selected models involved a modulation of either Bmal1 or Per2 transcription most likely by temperature or nutrient exposure cycles. This agreed with biological knowledge on temperature-dependent control of Per2 transcription. The strengths of systemic regulations were found to be significantly different according to mouse sex and genetic background.
8.4 CRN model reductions by tropical equilibrations
Participants: Eléonore Bellot, François Fages, Sylvain Soliman.
In the framework of the ANR-DFG SYMBIONT project we investigate mathematical justification of SEPI reductions based on Tikhonov's theorem and their computation using tropical algebra methods and constraint programming techniques 22.
This year we contributed to deliverable D2.1 on performance evaluation comparing polyhedral, SAT modulo theories (Z3) and constraint programming (BIOCHAM) methods.
8.5 Reduction of linear networks with separated timescales
Participants: Aurélien Naldi.
In 14, we introduced LNetReduce, a Python tool that simplifies linear dynamic networks, which are represented as digraphs labeled by integer timescale orders. Such models describe deterministic or stochastic monomolecular chemical reaction networks, but also random walks on weighted protein-protein interaction networks, spreading of infectious diseases and opinion in social networks, communication in computer networks. The reduced network is obtained by graph and label rewriting rules and reproduces the full network dynamics with good approximation at all timescales.
8.6 Buffering semantics for qualitative models
Participants: Aurélien Naldi.
In collaboration with colleagues in the discrete biomathematics group at FU Berlin and the I3S lab at université de Nice, we study extended Boolean models with intermediate variables representing unknown delays on the interactions. This approach eliminates implicit kinetic assumptions in the classical updating modes of these models, not only to recover relevant dynamical behaviours, but also to provide a framework for reasoning on kinetic constraints which are sufficient to enable these behaviours.
This year, preliminary results have been presented in two public talks and the results have been generalized. An article is in preparation.
8.7 Automated inference of Boolean models from molecular interaction maps using CaSQ
Participants: Martin Boutroux, Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
Molecular interaction maps have emerged as a meaningful way of representing biological mechanisms in a comprehensive and systematic manner. However, their static nature provides limited insights to the emerging behavior of the described biological system under different conditions. Computational modelling provides the means to study dynamic properties through in silico simulations and perturbations. Last year we described how to bridge the gap between static and dynamic representations of biological systems with CaSQ, a software tool that infers Boolean rules based on the topology and semantics of molecular interaction maps built with CellDesigner (see Section 7.1.2). We developed CaSQ by defining conversion rules and logical formulas for inferred Boolean models according to the topology and the annotations of the starting molecular interaction maps. We used CaSQ to produce executable files of existing molecular maps that differ in size, complexity and the use of SBGN standards. We also compared, where possible, the manually built logical models corresponding to a molecular map to the ones inferred by CaSQ. The tool is able to process large and complex maps built with CellDesigner (either following SBGN standards or not) and produce Boolean models in a standard output format, SBML-qual, that can be further analyzed using popular modelling tools. References, annotations and layout of the CellDesigner molecular map are retained in the obtained model, facilitating interoperability and model reusability. This year this was used in the context of the Covid 19 Disease Map project as a fundamental part of the pipeline 3
8.8 Setting the basis of best practices and standards for curation and annotation of logical models in biology
Participants: Aurélien Naldi, Anna Niarakis, Sylvain Soliman, Denis Thieffry.
The fast accumulation of biological data calls for their integration, analysis and exploitation through more systematic approaches. The generation of novel, relevant hypotheses from this enormous quantity of data remains challenging. Logical models have long been used to answer a variety of questions regarding the dynamical behaviours of regulatory networks. As the number of published logical models increases, there is a pressing need for systematic model annotation, referencing and curation in community-supported and standardised formats. In 12 we summarise the key topics and future directions of a meeting entitled ‘Annotation and curation of computational models in biology’, organised as part of the 2019 [BC]2 conference. The purpose of the meeting was to develop and drive forward a plan towards the standardised annotation of logical models, review and connect various ongoing projects of experts from different communities involved in the modelling and annotation of molecular biological entities, interactions, pathways and models. This article defines a roadmap towards the annotation and curation of logical models, including milestones for best practices and minimum standard requirements.On September 2021 a second workshop took part in Basel under the auspices of [BC]2 conference, to discuss the advancements in the logical modelling community and exchange with other modelling communitites of systems biology toward a common framework for annotated, accessible, reproducible and interoperable computational models in biology. Sylvain Soliman and Anna Niarakis were co-organizers and a the proceedings paper is under preparation.
In 8 , we review up-to-date efforts to address challenging biological questions by incorporating omic data into logic-based models and discuss critical difficulties in constructing and analyzing integrative, large-scale, logic-based models of biological mechanisms.Discrete, logic-based models are increasingly used to describe biological mechanisms. Initially introduced to study gene regulation, these models evolved to cover various molecular mechanisms, such as signaling, transcription factor cooperativity, and even metabolic processes. The abstract nature and amenability of discrete models to robust mathematical analyses make them appropriate for addressing a wide range of complex biological problems. Recent technological breakthroughs have generated a wealth of high-throughput data. Novel, literature-based representations of biological processes and emerging algorithms offer new opportunities for model construction.
In 6, we revise the work of the Community of Special Interest (COSI) in Computational Modelling of Biological Systems (SysMod) that was launched in 2016. SysMod’s main activity is an annual meeting at the Intelligent Systems for Molecular Biology (ISMB) conference, which brings together computer scientists, biologists, mathematicians, engineers, computational and systems biologists. SysMod maintains several online resources to facilitate interaction among the community members, including an online forum, a calendar of relevant meetings and a YouTube channel with talks and lectures of interest for the modelling community. For more than half a decade, the growing interest in computational systems modelling and multi-scale data integration has inspired and supported the SysMod community. Its members contribute to the annual COSI meeting and several related community workshops and meetings, focusing on computational modelling and standardisation efforts.
In 11 , we aim to unravel mechanisms governing the regulation of key transcription factors in RA and derive patient-specific models to gain more insights into the disease heterogeneity and the response to treatment. We first use publicly available transcriptomic datasets (peripheral blood) relative to RA and machine learning to create an RA-specific transcription factor (TF) co-regulatory network. The TF cooperativity network is subsequently enriched in signalling cascades and upstream regulators using a state-of-the-art, RA-specific molecular map. Then, the integrative network is used as a template to analyse patients’ data regarding their response to anti-TNF treatment and identify master regulators and upstream cascades affected by the treatment. Finally, we use the Boolean formalism to simulate in silico subparts of the integrated network and identify combinations and conditions that can switch on or off the identified TFs, mimicking the effects of single and combined perturbations.
8.9 Dynamical modelling of type I IFN responses in SARS-CoV2 infection
Participants: Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
In this work, we make use of the type I IFN graphical model developed during the COVID19 Disease Map project and available in the C19DM repository and the map-to-model translation framework (see Section 7.1.2), to obtain an executable, dynamic model of type I IFN signalling for in silico experimentation. Our goal is to understand how we can maximize the Antiviral Immune response (production of ISGs), limit Inflammation, and amplify the inhibition of viral replication through the production of the IFNs. Starting from the CellDesigner map of the type I Interferon signalling we use the tool CaSQ to obtain an SBML qual file with preliminary Boolean rules. To cope with specificities of the COVID19 Disease Map repository, the tool CaSQ was adapted (version 1.0.0) to include: a) automatic merging of identical SBML species that can appear for visual purposes on the XML CellDesigner graph, b) the active suffix in the species name when the dotted circle was used to denote activation of a certain species. The SBML qual file was processed to ensure compatibility regarding name display in GINsim, and also control nodes were added in an attempt to reduce the number of inputs (from 37 to 7) and ease the computational cost. Given the size of the model (given the number of nodes and edges), an exhaustive attractors search would not be feasible. Therefore, we searched for attractors for a given set of initial conditions that cover different biological scenarios of the type I IFN pathway with or without the infection, and in the presence or absence of drugs. For the analysis we used the CoLoMoTo notebook implementation. Besides the dynamical analysis of the model which is based on literature mining, we are also using the model to assess the impact of target nodes prioritized by omic data analysis and drug target enrichment, performing single and combined perturbations. An article is in preparation.
8.10 Fibroblasts as therapeutic targets in rheumatoid arthritis (RA) and cancer. Computational modeling of the metabolic reprogramming (glycolytic switch) in RA synovial fibroblasts (RASFs) and cancer associated fibroblasts (CAFs)
Participants: Sahar Aghakhani, Solene Mansour, Anna Niarakis, Sylvain Soliman.
Fibroblasts are critical regulators of several physiological processes linked to extracellular matrix regulation. Under certain conditions, fibroblasts can also transform into more aggressive phenotypes and contribute to disease pathophysiology. In this work, we highlight metabolic reprogramming as a critical event toward the transition of fibroblasts from quiescent to activated and aggressive cells, in rheumatoid arthritis and cancer. We draw obvious parallels and discuss how systems biology approaches and computational modeling could be employed to highlight targets of metabolic reprogramming and support the discovery of new lines of therapy. The objectives of this project are: a) First, to study similarities in the molecular and signalling pathways of RASFs and CAFs, which are involved in the regulation of glucose metabolism. b) Second, to create dynamic models that could be used for in silico perturbations in order to study the cells’ response to different conditions such as inflammation, hypoxia, and elevated ROS levels, to name a few. Dynamical modelling is indispensable if we want to understand the emergent behaviour of the cells when complex and intertwined pathways are involved. c) Lastly, to identify and propose a possible mechanism (common or not) that could explain the fibroblasts’ metabolic reprogramming regarding glucose in disease- specific conditions. An in-depth review focusing on RASFs and CAFs was recently published in Cancers 4.
8.11 Inference of a modular, large-scale Boolean network for modelling the Rheumatoid Arthritis fibroblast-like synoviocytes
Participants: Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
Rheumatoid arthritis fibroblast-like synoviocytes (RA FLS) are central players in the disease pathogenesis, as they are involved in the secretion of cytokines and proteolytic enzymes, exhibit invasive traits, high rate of self-proliferation and an apoptosis-resistant phenotype. Using the RA map and the tool CaSQ we build a large-scale dynamic model of RA FLS to study apoptosis, cell proliferation and growth, osteoclastogenesis and bone erosion, matrix degradation and cartilage destruction and inflammation outcomes. The RA FLS network was created selecting fibroblast-relevant sub-parts of the state-of-the-art RA map, and the Boolean rules were added using the tool CaSQ that infers logical formulae based on the topology and the semantics of the network (see Section 7.1.2). To cope with complexity, we also employ a “divide and conquer” strategy by creating separate executable sub-modules that comprise each only one phenotype. We present also reduced model versions that facilitate downstream analysis. Systematic testing of different initial conditions could further lead to predictions regarding the outcomes of specific perturbations, such as single or combined effects, simulated with the model by forcing or suppressing the activity of various factors of interest systematically. The goal is to gain a better understanding of the mechanisms that drive inflammation, resistance to apoptosis, high proliferation rate, and cartilage and bone degradation, and their coordination, to delineate and gain control of these outcomes (paper in preparation).
8.12 Data-enriched signalling networks to decipher the dual tumor promoting/supressing effects of the Syk tyrosine kinase
Participants: Aurélien Naldi.
Spleen tyrosine kinase (SYK) can behave as an oncogene or a tumor suppressor, depending on the cell and tissue type. As pharmacological SYK inhibitors are currently evaluated in clinical trials, it is important to gain more information on the molecular mechanisms underpinning these opposite roles. In 5, we reconstructed and compared data-enriched signaling networks using phosphoproteomic data from breast cancer and Burkitt lymphoma cell lines where SYK plays opposite roles. Our analysis shows that in breast cancer cells, the SYK target-enriched signaling pathways included intercellular adhesion and Hippo signaling components that are often linked to tumor suppression. In Burkitt lymphoma cells, the SYK target-enriched signaling pathways included molecules that could play a role in SYK pro-oncogenic function in B-cell lymphomas. These data demonstrate that proteomic profiling combined with mathematical network modeling allows untangling complex pathway interplays and revealing difficult to discern interactions among the SYK pathways that positively and negatively affect tumor formation and progression.
8.13 Computational model of the microtubule tyrosination cycle
Participants: François Fages, Jeremy Grignard, Sylvain Soliman.
Microtubules and their post-translational modifications involved in major cellular processes such as: mitosis, cardiomyocyte contraction, and neuronal differentiation. More precisely, in neurons, the post-translational modifications of detyrosination and tyrosination are crucial for neuronal plasticity, axon regeneration, recruitment and transports of proteins and correct neuronal wiring. We hypothesize that the decrease of density and length of microtubules and the loss of neuronal structures such as synapses, dendritic spine and growth cone which are correlated with the progressive cognitive decline may be the consequence of the dysregulation of the cycle detyrosination/tyrosination in neurodegenerative disorder. This hypothesis is investigated in collaboration with Servier by combining experimental approaches with mathematical modelling in a paper currently under review.
8.14 Computational model of inflammation process in skin
Participants: François Fages, Eléa Greugny, Mathieu Hemery.
Skin protects the body against external agents, for instance pathogens, irritants, or UV radiation, that can trigger inflammation. Inflammation is a complex phenomenon that is classified in two main types, acute and chronic. They are distinguished by different parameters such as the duration, the underlying mechanisms, the components involved like the type of immune cells, and the nature and intensity of the associated clinical signs. The computational models developed in collaboration with Johnson&Johnson France, combine mathematical and multi-agent modelling using BIOCHAM and EPISIM modelling tools.
In 7 we study the effect of pH on skin inflammation using a multi-agent model.
9 Bilateral contracts and grants with industry
9.1 Bilateral contracts with industry
9.1.1 Institut de Recherches Servier
Participants: François Fages, Jeremy Grignard, Sylvain Soliman.
In the framework of the Cifre PhD thesis of Jeremy Grignard at Servier, we work on the coupling between computational modeling and biological experiment design, and on chemical reaction network inference methods from data time series.
9.1.2 Johnson&Johnson Santé Beauté France
Participants: François Fages, Elea Greugny, Mathieu Hemery, Sylvain Soliman.
In the framework of the Cifre PhD thesis of Eléa Greugny at Johnson&Johnson Santé Beauté France, we work on the computational modeling of inflammatory process in the skin, using multi-scale modeling and multi-agent simulation.
9.1.3 IBM research, France
Participants: Marine Collery, François Fages, Julien Martinelli, Sylvain Soliman.
In the framework of the PhD thesis of Marine Collery at IBM France, we work on rule learning from time series data in the context of learning fraud detection rules in bank transactions.
10 Partnerships and cooperations
Participants: Eléonore Bellot, François Fages, Mathieu Hemery, Julien Martinelli, Sylvain Soliman.
10.1 International research visitors
10.1.1 Visits of international scientists
The following researchers have been invited for short visits:
The following researchers have been invited:
- Barbara Bravi, ENS Paris, for short visit and seminar;
- Zhang Xingjian, LIX, for a seminar.
10.2 National initiatives
10.2.1 ANR Projects
- ANR ifference on "Complexity theory with discrete ODEs", coordinated by Olivier Bournez, Ecole Polytechnique, with F. Fages (Inria), F. Chyzak (Inria), A. Durand Univ. Paris-Diderot, F. Madelaine P. Valarché Univ. Créteil, N20 Jerôme Durand-Lose, Orléans - Moulay Barkatou Thomas Cluzeau, Limoges - Mathieu Sablik, Toulouse
- ANR-DFG Symbiont (2018-2022) on “Symbolic Methods for Biological Systems”, coordinated by T. Sturm (CNRS, LORIA, Nancy, France) and A. Weber (Univ. Bonn, Germany) with F. Fages (Inria), F. Boulier (U. Lille), O. Radulescu (U. Montpellier), A. Schuppert (RWTH Aachen), S. Walcher (RWTH Aachen), W. Seiler (U. Kassel);
- ANR-MOST Biopsy (2016-2021) on “Biochemical Programming System”, coordinated by F. Molina (CNRS, Sys2diag, Montpellier) and J.H. Jiang (National Taiwan University), with F. Fages (Inria);
11 Dissemination
11.1 Promoting scientific activities
Participants: François Fages, Mathieu Hemery, Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
11.1.1 Scientific events: organisation
François Fages is member of the Steering Committee of the conference series Computational Methods for Systems Biology (CMSB) since 2008.
Anna Niarakis and Sylvain Soliman were co-organizers of the [BC]2 2021 Workshop “Toward a common framework for annotated, accessible, reproducible and interoperable computational models in biology”, September 13, 2021, Basel, Switzerland.
In addition, Anna Niarakis was co-organizer of the following events:
- SysMod Workshop 2021 Virtual;
- 6th Disease Maps Community Meeting, Virtual.
- Participation at the organizing committee of JOBIM 2021, 2022.
- Wellcome Trust Advanced Course in Computational Systems Biology for Complex Human Disease: from static to dynamic representations of disease mechanisms, Hinxton, Cambridge, UK 2021.
11.1.2 Scientific events: selection
François Fages was PC member of CMSB 2021, 19th International Conference on Computational Methods in Systems Biology, Bordeaux, France, 22nd-24th September 2021
Anna Niarakis was in the selection committee for SysMod 2021, JOBIM 2021, [BC]2 2021, WTAC 2021.
Sylvain Soliman was PC member of
- BIOINFORMATICS - BIOSTEC 2021, the International Joint Conference on Biomedical Engineering Systems and Technologies
- and CSBio 2021, The 12th International Conference on Computational Systems-Biology and Bioinformatics
Sylvain Soliman wrote reviews for CASC'21
11.1.3 Journal
François Fages is member of
- the Editorial Board of the Computer Science area of the Royal Society Open Science journal, since 2014;
- the Editorial Board of the journal RAIRO OR Operations Research, since 2004.
Anna Niarakis is member of the Editorial Board of Scientific Reports Nature Publishing.
She wrote reviews for Molecular Medicine, Plos Computational Biology, Immunoinformatics, Frontiers in Applied Mathematics and Statistics, Nucleic Acid Research and Cancers.
François Fages wrote reviews for Theoretical Computer Science and Bioinformatics.
Mathieu Hemery wrote reviews for Physical Review Research, Physical Review E and the Royal Society Open Science.
Sylvain Soliman wrote reviews for Bioinformatics and PLoS Computational Biology.
11.1.4 Invited talks
François Fages and Sylvain Soliman gave two invited talks at SIAM AG 2021, Sochi, August 2021.
Aurélien Naldi gave an invited online talk to the GT BIOSS, February 2021.
Anna Niarakis gave the following invited talks:
- Keynote Speaker, COMBINE 2021, virtual, Do you speak Systems Biology? Shaping a common language for community work, October 11th 2021
- Invited Lecture at the Summer School, Logical Modeling for Experimental Design in Current and Future Biotechnology and Biomedicine, Trondheim, NTNU, Norway, 17 August 2020 - 28 August 2021
- Invited Speaker, GREEKC COST ACTION Workshop, virtual, Introduction to Disease Maps: successes and challenges, 24 June of 2021
- Invited speaker, COVID-19 Disease Map, a computational knowledge repository of SARS-CoV-2 virus-host interaction mechanisms, MSM Viral Pandemics meetings, virtual, 28th of January 2021
11.1.5 Leadership within the scientific community
Anna Niarakis is:
- Member of the Scientific Advisory Board of REACTOME
- Co-leader of the Covid 19 Disease map project
- Co-leader of the Consortium Disease Maps
- Coordinator of SysMod (Systems Modelling)
11.1.6 Scientific expertise
François Fages evaluated one research project for UKRI BBSR, Biotechnology and Biological Sciences Research Council.
Anna Niarakis evaluated for:
- MRC Career Development Award
- Versus Arthritis UK PhD scholarship Award
- Wellcome Clinical Research Career Development Fellowship
She was also a member of the national jury for the concours CRCN INRAE 2021
11.1.7 Research administration
François Fages is member of
- "Bureau du Comité des équipes-projets du centre Inria SIF"
- "Bureau du département Informatique Données et Intelligence Artificielle (IDIA) de l'Institut Polytechnique de Paris"
- Scientific committee of the Doctorate School "ED 474 - Frontière de l'Innovation en Recherche (FIRE)" (from 2010 to October 2021)
- Ecole Doctorale ED IPP 626 ED, Institut Polytechnique de Paris, since its creation in 2019
Aurelien Naldi and Anna Niarakis are members of CoLoMoTo (Consortium for Logical Models and Tools).
Anna Niarakis is member of the French Bioinformatics Society.
Sylvain Soliman is member of the “Commission Scientifique” of Inria Saclay-IdF.
11.2 Teaching - Supervision - Juries
Participants: Eléonore Bellot, François Fages, Mathieu Hemery, Julien Martinelli, Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
11.2.1 Teaching
- International advanced course: Anna Niarakis, Ben Hall (co-organizers), Aurélien Naldi, Sylvain Soliman (trainers) Computational Systems Biology for Complex Human Disease, MRC University of Cambridge, Wellcome Trust course WGC Advanced Course, 13-17 December 2021, Hinxton, Cambridge UK.
- International advanced course: Aurélien Naldi, Anna Niarakis (trainers) Logical Modeling for Experimental Design in Biotechnology and Biomedicine, August 23-26 2021, NTNU, Trondheim, Norway.
- Master 2: François Fages (coordinator 24h and teacher 12h), Jérôme Feret (12h) C2-19 - Biochemical Programming, Master Parisien de Recherche en Informatique (MPRI), Paris.
- Ingénieur Ecole Polytechnique, Master 1: François Fages (coordinator, 18h lectures, 14h TD), Mathieu Hemery (6h TD) INF555 - Constraint-based Modeling and Algorithms for Decision Making ProblemsMaster Artificial Intelligence, Master Science and Technology, Ecole Polytechnique.
- Ingénieur Ecole Polytechnique, Master 1: François Fages, Sylvain Soliman (co-coordinators 60h) INF473L - Bioinformatics of interaction networks not taught in 2021
- Bachelor 3: François Fages (co-coordinator, 12h lectures), Mathieu Hemery (12h TD), CSE 307 - Constraint Logic Programming, Ecole Polytechnique.
- Bachelor 2: Eléonore Bellot (teacher 6h) CSE201 Object-oriented Programming in C++ TD and project supervision
- L2: Julien Martinelli (teaching assistant 42h) Real Analysis, Université de Paris / (teaching assistant 18h) Multivariate functions, Université de Paris
- Anna Niarakis is the pedagogical responsible for the Double Bachelor in Life Sciences and Informatics at Paris Saclay University.
11.2.2 Supervision
- PhD in progress: Sahar Aghakhani, “Fibroblasts as therapeutic targets in rheumatoid arthritis (RA) and cancer. Computational modeling of the metabolic reprogramming (glycolytic switch) in RA synovial fibroblasts (RASFs) and cancer associated fibroblasts (CAFs)” , ED Paris-Saclay, Sep. 2020, Anna Niarakis and Sylvain Soliman (50%)
- PhD stopped : Eléonore Bellot, “Réduction de modèles différentiels par résolution de contraintes d’algèbre tropicale (min,+)”, ED IPP, Ecole Polytechnique, Sept. 2018, F. Fages and S. Soliman (50-50%)
- PhD in progress : Marine Collery, “Apprentissage de règles à partir de données dépendantes du temps appliqué à la détection de fraudes”, ED IPP, Ecole Polytechnique, Sept. 2020, P. Bonnard, F. Fages (1/3) and R. Kuiters, IBM research France
- PhD in progress : Eléa Greugny, “Development and Implementation of a Mathematical Model of Inflammation in the Human Skin”, ED IPP, Ecole Polytechnique, Aug. 2019, F. Fages (1/3), J. Bensaci, G. Stamatas, Johnson&Johnson Santé Beauté France
- PhD in progress : Jérémy Grignard, “Apprentissage de modèles à partir de données pour la conception d’expériences de criblage et la recherche de médicaments”, ED IPP, Ecole Polytechnique, Mar. 2019, F. Fages and T. Dorval, Servier (50%)
- PhD in progress : Julien Martinelli, “Apprentissage de modèles mécanistes à partir de données temporelles, application à la personnalisation de la chronothérapie des cancers”, ED IPP, Ecole Polytechnique, Oct. 2018, F. Fages and A. Ballesta, INSERM (50%)
Anna Niarakis was a mentor for the 2021 iGem Evry -Paris Saclay team (gold medal).
11.2.3 Juries
François Fages participated in the juries of:
- HDR Anna Niarakis, Université d'Evry Paris-Saclay, Rapporteur, 10 mai 2021;
- PhD Gaspard Ducamp, Sorbonne Université, Rapporteur, 13 avril 2021;
- PhD Emilie Allart, Université de Lille, 29 janvier 2021.
Anna Niarakis participated in the juries of:
- PhD Nicolas Sompairac, "Unsupervised hierarchical deconvolution of gene expression data to unravel the tumor micro-environment complexity”, PSL, Paris, Institut Curie, December 10th, 2021;
- PhD Laure Talarmain, MRC Cancer Unit, Hutchison/MRC Research Centre, University of Cambridge, External Reviewer, February 4th, 2021
11.3 Popularization
Participants: Mathieu Hemery, Anna Niarakis.
11.3.1 Articles and contents
The Covid-19 disease map published in 18 is the subject of several popularization actions and web pages
Mathieu Hemery help the mediation team of the Saclay Center to realize a video of vulgarization around the notion of algorithm.
11.3.2 Education
Mathieu Hemery take part to the inria stand for the 30 years of the "Fête de la Science" on the campus of the École polytechnique.
Fête de la sciences : Genopole : visite guidée, oct. 2021.
12 Scientific production
12.1 Major publications
- 1 inproceedingsCompiling Elementary Mathematical Functions into Finite Chemical Reaction Networks via a Polynomialization Algorithm for ODEs.Proceedings of the nineteenth international conference on Computational Methods in Systems Biology,CMSB 2021 - 19th International Conference on Computational Methods in Systems BiologyBordeaux, FranceSeptember 2021
- 2 articleModel learning to identify systemic regulators of the peripheral circadian clock.Bioinformatics37Supplement_1July 2021, 9
- 3 articleCOVID19 Disease Map, a computational knowledge repository of virus-host interaction mechanisms.Molecular Systems Biology1710October 2021, e10387
12.2 Publications of the year
International journals
International peer-reviewed conferences
Conferences without proceedings
Reports & preprints