
Keywords
Computer Science and Digital Science
- A6.1. Methods in mathematical modeling
- A6.1.1. Continuous Modeling (PDE, ODE)
- A6.1.2. Stochastic Modeling
- A6.1.3. Discrete Modeling (multi-agent, people centered)
- A6.1.4. Multiscale modeling
- A6.2.1. Numerical analysis of PDE and ODE
- A6.2.3. Probabilistic methods
- A6.2.4. Statistical methods
- A6.3.1. Inverse problems
Other Research Topics and Application Domains
- B1.1.2. Molecular and cellular biology
- B1.1.3. Developmental biology
- B1.1.4. Genetics and genomics
- B1.1.5. Immunology
- B1.1.6. Evolutionnary biology
- B1.1.7. Bioinformatics
- B1.1.8. Mathematical biology
- B1.1.10. Systems and synthetic biology
- B2.2.1. Cardiovascular and respiratory diseases
- B2.2.3. Cancer
- B2.2.5. Immune system diseases
- B2.2.6. Neurodegenerative diseases
1 Team members, visitors, external collaborators
Research Scientists
- Mostafa Adimy [Team leader, INRIA, Senior Researcher, HDR]
- Samuel Bernard [CNRS, Researcher, HDR]
- Vincent Calvez [CNRS, Researcher, HDR]
- Fabien Crauste [CNRS, HDR]
- Olivier Gandrillon [CNRS, Senior Researcher, HDR]
- Thomas Lepoutre [INRIA, Researcher, HDR]
- Vitaly Volpert [CNRS, Senior Researcher, HDR]
Faculty Members
- Thibault Espinasse [UNIV LYON I, Associate Professor]
- Laurent Pujo-Menjouet [UNIV LYON I, Associate Professor, HDR]
- Leon Tiné [UNIV LYON I, Associate Professor]
Post-Doctoral Fellows
- Thi Nhu Thao Nguyen [Inria, from Feb 2022]
- Nicolas Torres [UNIV LYON I, from Apr 2022]
PhD Students
- Charlotte Dugourd [Inria and UNIV LYON I, from Oct 2022]
- Maxime Estavoyer [Inria and UNIV LYON I, from Feb 2022]
- Grégoire Ranson [UNIV LYON I and UNIV YORK]
Administrative Assistant
- Claire Sauer [INRIA]
2 Overall objectives
2.1 Presentation
Dracula is a joint research team between INRIA, Université Claude Bernard Lyon 1 (UCBL) and CNRS (Institut Camille-Jordan (ICJ, UMR 5208) and Laboratoire de Biologie et Modélisation de la Cellule (LBMC, UMR 5239)).
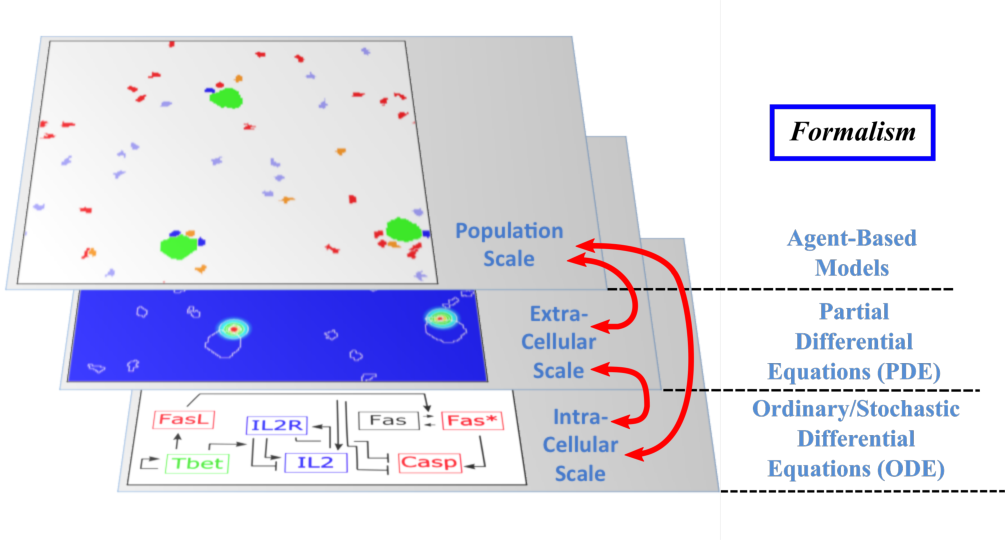
The Dracula project is devoted to multi-scale modeling in biology and medicine, and more specifically to the development of tools and methods to describe multi-scale processes in biology and medicine. Applications include normal and pathological hematopoiesis (for example leukemia), immune response, and other biological processes, like: tissue renewal, morphogenesis, atherosclerosis, prion disease, hormonal regulation of food intake, and so on. Multi-scale modeling implies simultaneous modeling of several levels of descriptions of biological processes: intra-cellular networks (molecular level), cell behavior (cellular level), dynamics of cell populations (organ or tissue) with the control by other organs (organism) (see Figure 1). Such modeling represents one of the major challenges in modern science due to its importance and because of the complexity of biological phenomena and of the presence of very different interconnected scales.
Although multi-scale modeling holds a great potential for biology and medicine, and despite the fact that a variety of techniques exists to deal with such problems, the complexity of the systems poses new challenges and needs the development of new tools. Moreover, different biological questions usually require different types of multi-scale modeling. The expected results of these studies are numerous. On one hand, they will shed new light on the understanding of specific biological and medical questions (for instance, what is the behavior of hematopoietic stem cells under pathological conditions? Or how to efficiently stimulate an immune response in order to design new vaccines?). On the other hand, the modeling methods developed here for specific processes are relevant to study other complex biological systems. We pay a special attention on developing methods that are not restricted to one or two applications.
An important part of our researches is performed in close collaboration with biologists and physicians in order to stay in contact with the biological and medical goals. The presence, within the project, of a biologist (Olivier Gandrillon) who has acquired over the years the know-how required for interacting with mathematicians is probably one of the main assets of the project. He participates actively in many tasks of our program, stimulates interactions between members of the project and biologists, and everyone benefits from his expertise in molecular and cell biology.
2.2 Keywords
Multi-scale modeling; Hybrid modeling; Mathematical Biology; Computational Biology; Immune response modeling; Normal and pathological hematopoiesis; Multi-scale cancer modeling; Regulatory networks; Reaction-diffusion equation; Structured partial differential equations; Delay differential equations; Agent-based modeling; Dynamical systems.
2.3 Research axis 1: Mathematical modeling for cell population dynamics
Executive summary
Stem cells are essential for development and keep the maintenance of many tissues homeostasis. They are characterized by their ability to self-renew as well as to produce differentiated cells. They vary enormously, for each organ, in their proliferation capacity, their potency to produce different cell lineage and their response to various environmental cues. How a cell will react to a given external signal does not depend only on its current state but also on its environment. Understanding the effect of cell-to-cell heterogeneity and the spatial organization of cell populations is therefore necessary to help keeping the normal function of an organ.
We develop mathematical tools and methods to study cell population dynamics and other biological processes: stability of steady sates, existence of bifurcations, kinetic properties, spatial organization, in finely detailed cell populations. The main tools we use are hybrid discrete-continuous models, reaction-diffusion equations, structured models (in which the population is endowed with relevant structures or traits), delay differential systems, agent-based models. Our team has acquired an international expertise in the fields of analysis of reaction-diffusion and structured equations, particularly integro-differential and delay differential equations.
The mathematical methods we develop are not restricted to hematopoietic system (Research axis 2), and immune response (Research axis 3), rather we apply them in many other biological phenomena, for example: tissue renewal, morphogenesis, prion disease, atherosclerosis, hormonal regulation of food intake, cancer, and others.
Project-team positioning
The focus of this objective is the development, analysis and application of hybrid discrete-continuous, reaction-diffusion and structured partial differential models. The structured equations allow a fine description of a population as some structures (age, maturity, intracellular content) change with time. In many cases, structured equations can be partially integrated to yield integro-differential equations (ordinary or partial differential equations involving non-local integral terms), time-delay differential or time-delay partial differential, or coupled differential-difference models. Analysis of integro-differential and time-delay systems deals with existence of solutions and their stability. Applications are found in the study of normal and pathological hematopoietic system (Research axis 2), immune response (Research axis 3), morphogenesis, prion disease, cancer development and treatment, and generally in tissue renewal problems. Models based on structured equations are especially useful to take into account the effect of finite time cells take to divide, die or become mature. Reaction-diffusion equations are used in order to describe spatial distribution of cell populations. It is a well developed area of research in our team which includes qualitative properties of travelling waves for reaction-diffusion systems with or without delay, and complex nonlinear dynamics.
Our team has developed a solid expertise in mathematical analysis of reaction-diffusion with or without delay and structured equations (in particular, delay differential equations) and one of the most prolific. Other major groups are the teams of Benoit Perthame (Pierre et Marie CURIE University and Mamba, Paris), Emmanuel Grenier (Ecole normale supérieure de Lyon and NUMED), Odo Diekmann (Utrecht University, The Netherlands), Avner Friedman (The Ohio State University, USA), Jianhong Wu (York University, Canada), Glenn Webb (Vanderbilt University, Nashville, USA), Philip K. Maini (University of Oxford, England), Mark Chaplain (University of St Andrews, Scotland), Nicola Bellomo (University of Turin, Italy). Most of the members of all these groups and of our team belong to the same mathematical community working on partial differential equations and dynamical systems with applications to biology and medicine.
Collaborations
- University of Toronto, Canada; Mathematical analysis and applications of reaction-diffusion equations (more than 30 joint papers).
- Institute of Problems of Mechanical Engineering, St.Petersburg, Russia; Dynamics of cell renewal (more than 10 joint papers).
- Department of Cell and Molecular Biology and Department of Forensic Medicine, Stockholm, Sweden; Dynamics of cell generation and turnover (3 joint papers).
- Universities of Tlemcen (Algeria) and Marrakech (Morocco); Delay differential equations (7 joint papers)
2.4 Research axis 2: Multi-scale modeling of hematopoiesis and leukemia
Executive summary
Hematopoiesis is a complex process that begins with hematopoietic stem cells (HSCs) and results in formation of mature cells: red blood cells, white cells and platelets. Blood cells are produced in the bone marrow, from where mature cells are released into the blood stream. Hematopoiesis is based on a balance between cell proliferation (including self-renewal), differentiation and apoptosis. The choice between these three possibilities is determined by intra-cellular regulatory networks and by numerous control mechanisms in the bone marrow or carried out by other organs. Intra-cellular regulatory networks are complex biochemical reactions involving proteins, enzymes and signalling molecules. The deregulation of hematopoiesis can result in numerous blood diseases including leukemia (a cancer of blood cells). One important type of leukemia is Chronic Myeloid Leukemia (CML). The strong tyrosine kinase activity of the BCR-ABL protein is the basis for the main cell effects that are observed in CML: significant proliferation, anti-apoptotic effect, disruption of stroma adhesion properties. This explains the presence in CML blood of a very important number of cells belonging to the myeloid lineage, at all stages of maturation.
Multi-scale modeling in hematopoiesis holds a great potential. A variety of techniques exists to deal with this problem. However, the complexity of the system poses new difficulties and leads to the development of new tools. The expected results of this study are numerous. On one hand, it will shed new light on the different physiological mechanisms that converge toward the continuous regeneration of blood cells, for example: the understanding of deregulation of erythropoiesis (the process of red blood cell production) under drug treatments (this can lead to lack of red blood cells (anemia), or a surplus of red blood cells), the dynamic of leukemic cells under the action of drugs and the control of their resistance to these treatments.
Project team positioning
Multi-scale modeling of hematopoiesis is one of the key points of the project that has started in the early stage of the Dracula team. Investigated by all the team members, it took many years of close discussion with biologists to get the best understanding of the key role played by the most important molecules, hormones, kinase cascade, cell communication up to the latest knowledge. One of the important questions here is to identify particular biological mechanisms (intracellular regulation, control mechanisms) and to integrate them in the different models. Our main work consisted in the development of a hybrid (continuous/discrete) model for red blood cell progenitor proliferation, survival/death, differentiation, and migration. Cells are modeled as discrete objects, and the extracellular medium is described by continuous equations for extracellular concentrations. This is to our knowledge the most complete model for erythropoiesis to date, and the only one using a multi-scale formalism. Other models published by our group and others for hematopoiesis are population-based models, mostly population structured equations (transport partial differential equations or delay differential equations). The interest in modeling hematopoiesis dates back to the 70's and two groups have been responsible for most of development in the past 40 years: Markus Loeffer's team in Leipzig, Germany (Wichmann et al. 1976, in Mathematical Models in Medicine) and Michael Mackey's team at McGill University, Montreal, Canada (Mackey 1978, Blood). Our model differs from population based models in that the regulation is directly modeled at the molecular level (See Figure 1) rather than acting on rates at the population level. Thus we can take into account non-predictable effects of interactions between different molecular pathways and between cells that would otherwise be lost in the global population rates.
Regarding modeling leukemia, we concentrated on Chronic Myeloid Leukemia (CML) and its treatment. We considered models based on ordinary differential equations for the action of the main proteins involved in CML (as BCR-ABL protein), and of transport equations (with or without delay, physiologically structured or not) to represent healthy and leukemic cell populations, take into account many interactions between proteins (especially BCR-ABL), cells (anti-apoptotic effect, etc.). The development of models for CML allowed us to interact with Franck Nicolini in Lyon (Centre Hospitalier de Lyon) and Doron Levy (Maryland University). Different schools developed models for CML and its treatment. The three leading groups are the ones of Franziska Michor (Harvard School of public health), Ingo Roeder (Institute for Medical Informatics and Biometry, Dresden) and Michael Mackey (McGill University).
Collaborations
Members of the team have worked for several years in collaboration with biologists (François Morlé, University Lyon 1) and hematologists (Charles Dumontet, Lyon and Mark Koury, Nashville) on the Modelling of normal and pathological hematopoiesis .
The work on modeling Leukemia is based on two major collaborations: firstly, an ongoing (since 2011) mathematical collaboration with the University of Maryland through the program Associate Teams Inria project, “Modelling Leukemia”. Secondly, an ongoing (since 2012) collaboration with a clinician from Hospices Civils de Lyon (Dr. F.E. Nicolini). In this framework, we shall have soon access to the data of the clinical trial PETALs ( patients).
2.5 Research axis 3: Multi-scale modeling of the immune response
Executive summary
Vaccination represents a worldwide health, social and economical challenge as it has allowed the eradication or the strong containment of several devastating diseases over the past century. However to date, most of the effective vaccines rely on the generation of neutralizing antibody responses and such vaccines have proven largely unsuccessful in the prevention against some pathogens, such as HIV or malaria. In such cases, vaccines geared towards the generation of CD8 T cell immunity may provide a better protection. The generation of memory CD8 T cells following antigenic immunization is a long process (lasting up to month in murine preclinical models), therefore strongly slowing the process of vaccine monitoring in preclinical studies. Thus, the dynamical modeling of the CD8 T cell immune response both at the cellular and molecular levels should provide an important tool to better understand the dynamics of the response and to speed-up the process and reduce costs of vaccine development.
However, currently published cellular models of the immune response are either over-simplified, not predicting important parameters of this response, or too complicated for most of their parameters to be accessible for experimental measurements, thus impeding their biological validation. Dynamical models of the CD8 T cell response at the molecular level are very scarce and there is no multi-scale model of the immune response giving insights into both the regulation at the molecular scale and the consequences on cell population dynamics.
The objective of this research axis is therefore to develop a predictive multi-scale model of the CD8 T cell response, by confronting the model at different stages to in vivo-acquired experimental data, in order to be able to investigate the influence of early molecular events on cell population dynamics few days or weeks later.
Project-team positioning
We are aiming at building and analyzing a multi-scale model of the CD8 T cell immune response, from the molecular to the cellular and potentially organismal scale. This consists in describing the dynamics at each scale with relevant formalisms as well as the careful description of the couplings between scales.
Only few research groups are actually working on the CD8 T cell immune response around the world, and none of them deals with multi-scale modeling of this response. A network developed around Alan Perelson's work in theoretical immunology in the last decades, at Los Alamos National Laboratory, and involves mainly people in various US universities or institutes. In Europe, Rob De Boer's group of theoretical immunology in Utrecht, Netherlands, is the historical leader in the CD8 T cell dynamics modeling. We considered the models developed in these groups when we started our project, and we contributed to improve them by using nonlinearities accounting for cell population interactions to regulate the response. Also, our initial focus was on the generation of memory cells associated with vaccine development so we modeled CD8 T cell responses against influenza and vaccinia viruses, whereas other groups usually consider LCMV in its chronic form.
Ron Germain's group at the NIH, and Grégoire Altan-Bonnet in subsequent works, focused on the molecular regulation of the CD4 and CD8 T cell immune responses. In particular, they built the Simmune software, which allows the modeling and simulation of molecular interactions. This software is not really devoted to multi-scale modeling yet it provides an interesting tool to describe molecular interactions. Since our aim is to couple molecular and cellular scales at the tissue level, and we do not want to consider large networks but rather small-simplified informative interaction networks, we are confident that our approach is complementary of these works.
Within Inria project-teams, NUMED develops multi-scale approaches for biological problems, and MAMBA and MONC mention models of cancer progression and treatment including immune responses. In the first case the methodology is similar, and collaborations between NUMED and DRACULA already exist (both teams are located in Lyon), but applications differ. In the second case, MAMBA and MONC are mainly focused on cancer modeling and up to now are motivated by including an action of the immune system in the fight against cancer, which is very different from what we are developing. However, both modeling approaches are complementary and could lead to interactions, in particular in the light of recent advances in medical research pointing towards an important role - and high expectations - of the immune reaction in fighting cancers. Finally, SISTM also focuses on the modeling of the immune response, mainly against HIV, but the motivation is very similar to ours: the objective is to provide tools and methods in order to efficiently develop vaccines. They consider the CD4 T cell response instead of the CD8 T cell response, and biostatistics to achieve their goals instead of multi-scale models, yet even though there is no interaction between SISTM and DRACULA at this moment our methods and objectives are close enough to foreshadow future collaborations.
Collaborations
On this topic our main collaborators are members of Jacqueline Marvel's team in Lyon in the CIRI (Centre International de Recherche en Infectiologie INSERM U1111): Dr. Jacqueline Marvel, head of the team, Dr. Christophe Arpin (CR CNRS), and other technicians and engineers of the team. They are all immunologists, specialists of the CD8 T cell response and of the generation of memory CD8 T cells.
We also interact with private companies: AltraBio, that provides tools for data analysis, and CosmoTech, that develops a modeling and simulating platform that should allow transferring our model on an easy-to-use platform devoted to commercial uses.
2.6 Evolution of research direction during the last evaluation
Reminder of the objectives given for the last evaluation
The aim of this project is the development of modern tools for multi-scale modeling in biological phenomena. During the period 2014-2017, the objectives we had fixed were to develop modern tools for multi-scale modeling of biological phenomena, as detailed hereafter:
- Multi-scale modeling of erythropoiesis, the process of red blood cell production, in order to describe normal, stress, and pathological erythropoiesis, using mathematical and computational models. This led to:
- Multi-scale modeling of the CD8 T cell immune response, in order to develop a predictive model of the CD8 T cell response, by confronting the model at different stages to in vivo-acquired experimental data;
- Population dynamics modeling, with the aim to develop general mathematical tools to study them. The main tools we were using were structured equations, in which the cell population is endowed with relevant structures, or traits. We identified limitations in using these formalisms, this is why we started developing multi-scale approaches;
- Modeling of Chronic Myeloid Leukemia (CML) treatment, using ordinary differential equations models. Our team had already developed a first model of mutant leukemic cells being resistant to chemotherapy. A next step would be to identify the parameters using experimental data;
- Multi-scale modeling carried out on the basis of hybrid discrete-continuous models, where dissipative particle dynamics (DPD) are used in order to describe individual cells and relatively small cell populations, partial differential equations (PDE) are used to describe concentrations of bio-chemical substances in the extracellular matrix, and ordinary differential equations for intracellular regulatory networks (Figure 1). An emphasis would be made on developing codes that are both flexible and powerful enough to implement variants of the model, perform simulations, produce desired outputs, and provide tools for analysis; to do so:
- We planned to contribute to a recent project named chronos, whose code (written in C++) represents heterogeneous populations of individual cells evolving in time and interacting physically and biochemically, and the objective is to make the code flexible enough to implement different formalisms within the same model, so that different components of the model can be represented in the most appropriate way;
- Partial differential equations (PDE) analysis, with a focus on reaction-diffusion equations, transport equations (hyperbolic PDEs) in which the structure can be age, maturity, protein concentration, etc., with particular cases where transport equations are reduced to delay differential equations (DDE).
Comments on these objectives over the evaluation period
We have had strong contributions to objectives 1, 2, 3, 4, and consequently to objective 5, as well as to objective 7, as mentioned in previous sections. These contributions represented the core of the team's research activity over the evaluation period, as stressed by our publications. It is however noticeable that multi-scale modeling of the immune response and of pathological hematopoiesis (leukemia) has come to represent a proportionally more important part of our activity.
Objective 6 has been pursued, the project chronos evolved to a better defined project SiMuScale that is currently being developed and aims at structuring the team's activity and providing a simulation platform that could be adapted to various biological questions necessitating multi-scale modeling.
Objectives for the next four years
The main objectives for the next four years are to continue to improve the 3 previous points: 1) Mathematical and computational modeling for cell population dynamics; 2) Multi-scale modeling of hematopoiesis and leukemia; 3) Multi-scale modeling of the immune response. In addition, we will pursue our effort to develop a simulation platform for multi-scale models (SiMuScale) and we intend to develop the use of mixed effect models and other statistical approaches to deal with the challenges offered by modern biology, in particular the generation of single cell data.
3 Research program
3.1 Mixed-effect models and statistical approaches
Most of biological and medical data that our team has to deal with consist in time series of experimental measurements (cell counts, gene expression level, etc.). The intrinsic variability of any biological system complicates its confrontation to models. The trivial use of means, eliminating the data variance, is but a second-best solution. Furthermore, the amount of data that can be experimentally generated often limits the use of classical mathematical approaches because model’s identifiability or parameter identifiability cannot be obtained. In order to overcome this issue and to efficiently take advantage of existing and available data, we plan to use mixed effect models for various applications (for instance: leukemia treatment modeling, immune response modeling). Such models were initially developed to account for individual behaviors within a population by characterizing distributions of parameter values instead of a unique parameter value. We plan to use those approaches both within that frame (for example, taking into account longitudinal studies on different patients, or different mice) but also to extend its validity in a different context: we will consider different ex vivo experiments as being “different individuals”: this will allow us to make the most of the experience-to-experience variations.
Such approaches need expertise in statistics to be correctly implemented, and we will rely on the presence of Céline Vial in the team to do so. Céline Vial is an expert in applied statistics and her experience already motivated the use of better statistical methods in various research themes. The increasing use of single cell technologies in biology make such approaches necessary and it is going to be critical for the project to acquire such skills.
3.2 Development of a simulation platform
We have put some effort in developing the SiMuScale platform, a software coded in dedicated to exploring multiscale population models, since 2014. In order to answer the challenges of multi-scale modeling it is necessary to possess an all-purpose, fast and flexible modeling tool, and SiMuScale is the choice we made. Since it is based on a core containing the simulator, and on plug-ins that contain the biological specifications of each cell, this software will make it easier for members of the team – and potentially other modelers – to focus on the model and to capitalize on existing models, which all share the same framework and are compatible with each other. Within the next four years, SiMuScale should be widely accessible and daily used in the team for multi-scale modeling. It will be developed into a real-case context, the modeling of the hematopoietic stem cell niche, in collaboration with clinicians (Eric Solary, INSERM) and physicists (Bertrand Laforge, UPMC).
3.3 Mathematical and computational modeling
Multi-scale modeling of hematopoiesis is one of the key points of the project that has started in the early stage of the Dracula team. Investigated by the team members, it took many years of close discussion with biologists to get the best understanding of the key role played by the most important molecules, hormones, kinase cascade, cell communication up to the latest knowledge. An approach that we used is based on hybrid discrete-continuous models, where cells are considered as individual objects, intracellular regulatory networks are described with ordinary differential equations, extracellular concentrations with diffusion or diffusion-convection equations (see Figure 1). These modeling tools require the expertise of all team members to get the most qualitative satisfactory model. The obtained models will be applied particularly to describe normal and pathological hematopoiesis as well as immune response.
3.4 From hybrid dynamics to continuum mechanics
Hybrid discrete-continuous methods are well adapted to describe biological cells. However, they are not appropriate for the qualitative investigation of the corresponding phenomena. Therefore, hybrid model approach should be combined with continuous models. If we consider cell populations as a continuous medium, then cell concentrations can be described by reaction-diffusion systems of equations with convective terms. The diffusion terms correspond to a random cell motion and the reaction terms to cell proliferation, differentiation and death. We will continue our studies of stability, nonlinear dynamics and pattern formation. Theoretical investigations of reaction-diffusion models will be accompanied by numerical simulations and will be applied to study cell population dynamic.
3.5 Structured partial differential equations
Hyperbolic problems are also of importance when describing cell population dynamics. They are structured transport partial differential equations, in which the structure is a characteristic of the considered population, for instance age, size, maturity, etc. In the scope of multi-scale modeling, protein concentrations as structure variables can precisely indicate the nature of cellular events cells undergo (differentiation, apoptosis), by allowing a representation of cell populations in a multi-dimensional space. Several questions are still open in the study of this problem, yet we will continue our analysis of these equations by focusing in particular on the asymptotic behavior of the system (stability, oscillations) and numerical simulations.
3.6 Delay differential equations
The use of age structure in PDE often leads to a reduction (by integration over the age variable) to delay differential equations. Delay differential equations are particularly useful for situations where the processes are controlled through feedback loops acting after a certain time. For example, in the evolution of cell populations the transmission of control signals can be related to some processes as division, differentiation, maturation, apoptosis, etc. Delay differential equations offer good tools to study the behavior of the systems. Our main investigation will be the effect of perturbations of the parameters, as cell cycle duration, apoptosis, differentiation, self-renewal, etc., on the behavior of the system, in relation for instance with some pathological situations. The mathematical analysis of delay differential equations is often complicated and needs the development of new criteria to be performed.
3.7 Multi-scale modeling of the immune response
The main objective of this part is to develop models that make it possible to investigate the dynamics of the adaptive CD8 T cell immune response, and in particular to focus on the consequences of early molecular events on the cellular dynamics few days or weeks later: this would help developing predictive tools of the immune response in order to facilitate vaccine development and reduce costs. This work requires a close and intensive collaboration with immunologist partners.
We recently published a model of the CD8 T cell immune response characterizing differentiation stages, identified by biomarkers, able to predict the quantity of memory cells from early measurements, 38. In parallel, we improved our multiscale model of the CD8 T cell immune response, by implementing a full differentiation scheme, from naïve to memory cells, based on a limited set of genes and transcription factors.
Our first task will be to infer an appropriate gene regulatory network (GRN) using single cell data analysis (generate transcriptomics data of the CD8 T cell response to diverse pathogens), the previous biomarkers we identified and associated to differentiation stages, as well as piecewise-deterministic Markov processes (Ulysse Herbach's PhD thesis, ongoing).
Our second task will be to update our multiscale model by first implementing the new differentiation scheme we identified 38, and second by embedding CD8 T cells with the GRN obtained in our first task (see above). This will lead to a multi-scale model incorporating description of the CD8 T cell immune response both at the molecular and the cellular levels (Simon Girel's PhD thesis, ongoing).
In order to further develop our multiscale model, we will consider an agent-based approach for the description of the cellular dynamics. Yet, such models, coupled to continuous models describing GRN dynamics, are computationally expensive, so we will focus on alternative strategies, in particular on descriptions of the cellular dynamics through both continuous and discrete models, efficiently coupled. Using discrete models for low cell numbers and continuous (partial differential equations) models for large cell numbers, with appropriate coupling strategies, can lead to faster numerical simulations, and consequently can allow performing intense parameter estimation procedures that are necessary to validate models by confronting them to experimental data, both at the molecular and cellular scales.
The final objective will be to capture CD8 T cell responses in different immunization contexts (different pathogens, tumor) and to predict cellular outcomes from molecular events.
3.8 Dynamical network inference from single-cell data
Up to now, all of our multiscale models have incorporated a dynamical molecular network that was build “by hand” after a thorough review of the literature. It would be highly valuable to infer it directly from gene expression data. However, this remains very challenging from a methodological point of view. We started exploring an original solution for such inference by using the information contained within gene expression distributions. Such distributions can be acquired through novel techniques where gene expression levels are quantified at the single cell level. We propose to view the inference problem as a fitting procedure for a mechanistic gene network model that is inherently stochastic and takes not only protein, but also mRNA levels into account. This approach led to very encouraging results 39 and we will actively pursue in that direction, especially in the light of the foreseeable explosion of single cell data.
3.9 Leukemia modeling
Imatinib and other tyrosine kinase inhibitors (TKIs) have marked a revolution in the treatment of Chronic Myelogenous Leukemia (CML). Yet, most patients are not cured, and must take their treatment for life. Deeper mechanistic understanding could improve TKI combination therapies to better control the residual leukemic cell population. In a collaboration with the Hospital Lyon Sud and the University of Maryland, we have developed mathematical models that integrate CML and an autologous immune response (35, 36 and 37). These studies have lent theoretical support to the idea that the immune system plays a rôle in maintaining remission over long periods. Our mathematical model predicts that upon treatment discontinuation, the immune system can control the disease and prevent a relapse. There is however a possibility for relapse via a sneak-though mechanism 35. Research in the next four years will focus in the Phase III PETALS trial. In the PETALS trial, the second generation TKI Nilotinib is combined with Peg-IFN, an interferon that is thought to enhance the immune response. We plan to: 1) Adapt the model to take into account the early dynamics (first three months). 2) Use a mixed-effect approach to analyse the effect of the combination, and find population and individual parameters related to treatment efficacy and immune system response. 3) Optimise long-term treatment strategies to reduce or cease treatment and make personalised predictions based on mixed-effect parameters, to minimise the long-term probability of relapse.
4 Application domains
See the previous sections.
5 New results
Participants: [ All team members.
5.1 Single-cell, genetic regulation network and cell differentiation
5.1.1 Reverse engineering of a mechanistic model of gene expression using metastability and temporal dynamics
Differentiation can be modeled at the single cell level as a stochastic process resulting from the dynamical functioning of an underlying Gene Regulatory Network (GRN), driving stem or progenitor cells to one or many differentiated cell types. Metastability seems inherent to differentiation process as a consequence of the limited number of cell types. Moreover, mRNA is known to be generally produced by bursts, which can give rise to highly variable non-Gaussian behavior, making the estimation of a GRN from transcriptional profiles challenging. In the paper 26 (see also the thesis 29), we present CARDAMOM (Cell type Analysis from scRna-seq Data achieved from a Mixture MOdel), a new algorithm for inferring a GRN from timestamped scRNA-seq data, which crucially exploits these notions of metastability and transcriptional bursting. We show that such inference can be seen as the successive resolution of as many regression problem as timepoints, after a preliminary clustering of the whole set of cells with regards to their associated bursts frequency. We demonstrate the ability of CARDAMOM to infer a reliable GRN from in silico expression datasets, with good computational speed. To the best of our knowledge, this is the first description of a method which uses the concept of metastability for performing GRN inference.
5.1.2 The best of both worlds: Combining population genetic and quantitative genetic models
Numerous traits under migration-selection balance are shown to exhibit complex patterns of genetic architecture with large variance in effect sizes. However, the conditions under which such genetic architectures are stable have yet to be investigated, because studying the influence of a large number of small allelic effects on the maintenance of spatial polymorphism is mathematically challenging, due to the high complexity of the systems that arise. In particular, in the most simple case of a haploid population in a two-patch environment, while it is known from population genetics that polymorphism at a single major-effect locus is stable in the symmetric case, there exists no analytical predictions on how this polymorphism holds when a polygenic background also contributes to the trait. In 14 (see also the thesis 27), we propose to answer this question by introducing a new eco-evo methodology that allows us to take into account the combined contributions of a major-effect locus and of a quantitative background resulting from small-effect loci, where inheritance is encoded according to an extension to the infinitesimal model. In a regime of small variance contributed by the quantitative loci, we justify that traits are concentrated around the major alleles, according to a normal distribution, using new convex analysis arguments. This allows a reduction in the complexity of the system using a separation of time scales approach. We predict an undocumented phenomenon of loss of polymorphism at the major-effect locus despite strong selection for local adaptation, because the quantitative background slowly disrupts the rapidly established polymorphism at the major-effect locus, which is confirmed by individual-based simulations. Our study highlights how segregation of a quantitative background can greatly impact the dynamics of major-effect loci by provoking migrational meltdowns. We also provide a comprehensive toolbox designed to describe how to apply our method to more complex population genetic models.
5.1.3 Hematopoietic differentiation is characterized by a transient peak of entropy at a single-cell level
Background: Mature blood cells arise from hematopoietic stem cells in the bone marrow by a process of differentiation along one of several different lineage trajectories. This is often represented as a series of discrete steps of increasing progenitor cell commitment to a given lineage, but as for differentiation in general, whether the process is instructive or stochastic remains controversial. In 15, we examine this question by analyzing single-cell transcriptomic data from human bone marrow cells, assessing cell-to-cell variability along the trajectories of hematopoietic differentiation into four different types of mature blood cells. The instructive model predicts that cells will be following the same sequence of instructions and that there will be minimal variability of gene expression between them throughout the process, while the stochastic model predicts a role for cell-to-cell variability when lineage commitments are being made. Results: Applying Shannon entropy to measure cell-to-cell variability among human hematopoietic bone marrow cells at the same stage of differentiation, we observed a transient peak of gene expression variability occurring at characteristic points in all hematopoietic differentiation pathways. Strikingly, the genes whose cell-to-cell variation of expression fluctuated the most over the course of a given differentiation trajectory are pathway-specific genes, whereas genes which showed the greatest variation of mean expression are common to all pathways. Finally, we showed that the level of cell-to-cell variation is increased in the most immature compartment of hematopoiesis in myelodysplastic syndromes. Conclusions: These data suggest that human hematopoietic differentiation could be better conceptualized as a dynamical stochastic process with a transient stage of cellular indetermination, and strongly support the stochastic view of differentiation. They also highlight the need to consider the role of stochastic gene expression in complex physiological processes and pathologies such as cancers, paving the way for possible noise-based therapies through epigenetic regulation.
5.1.4 CD8 memory precursor cell generation is a continuous process
In the paper 25, we studied the generation of memory precursor cells following an acute infection by analyzing single-cell RNA-seq data that contained CD8 T cells collected during the postinfection expansion phase. We used different tools to reconstruct the developmental trajectory that CD8 T cells followed after activation. Cells that exhibited a memory precursor signature were identified and positioned on this trajectory. We found that these memory precursors are generated continuously with increasing numbers arising over time. Similarly, expression of genes associated with effector functions was also found to be raised in memory precursors at later time points. The ability of cells to enter quiescence and differentiate into memory cells was confirmed by BrdU pulse-chase experiment in vivo. Analysis of cell counts indicates that the vast majority of memory cells are generated at later time points from cells that have extensively divided.
5.1.5 Theoretical evidence of osteoblast self-inhibition after activation of the genetic regulatory network controlling mineralization
Bone is a hard-soft biomaterial built through a self-assembly process under genetic regulatory network (GRN) monitoring. The paper 12 aims to capture the behavior of the bone GRN part that controls mineralization by using a mathematical model. Here, we provide an advanced review of empirical evidence about interactions between gene coding (i) transcription factors and (ii) bone proteins. These interactions are modeled with nonlinear differential equations using Michaelis-Menten and Hill functions. Compared to empirical evidence - coming from osteoblasts culture -, the two best systems use factors of inhibition from the start of the activation of each gene. It reveals negative indirect interactions coming from either negative feedback loops or the recently depicted micro-RNAs. The difference between the two systems also lies in the BSP equation and two ways for activating and reducing its production. Thus, it highlights the critical role of BSP in the bone GRN that acts on bone mineralization. Our study provides the first theoretical evidence of osteoblast self-inhibition after activation of the genetic regulatory network controlling mineralization with this work.
5.2 Viral replication in cell culture
5.2.1 Infection spreading in cell culture as a reaction-diffusion wave
Infection spreading in cell culture occurs due to virus replication in infected cells and its random motion in the extracellular space. Multiplicity of infection experiments in cell cultures are conventionally used for the characterization of viral infection by the number of viral plaques and the rate of their growth. In 7, we describe this process with a delay reaction-diffusion system of equations for the concentrations of uninfected cells, infected cells, virus, and interferon. Time delay corresponds to the duration of viral replication inside infected cells. We show that infection propagates in cell culture as a reaction-diffusion wave, we determine the wave speed and prove its existence. Next, we carry out numerical simulations and identify three stages of infection progression: infection decay during time delay due to virus replication, explosive growth of viral load when infected cells begin to reproduce it, and finally, wave-like infection progression in cell culture characterized by a constant or slowly growing total viral load. The modelling results are in agreement with the experimental data for the coronavirus infection in a culture of epithelial cells and for some other experiments. The presence of interferon produced by infected cells decreases the viral load but does not change the speed of infection progression in cell culture. In the 2D modelling, the total viral load grows faster than in the 1D case due to the increase of plaque perimeter.
5.2.2 Viral infection spreading and mutation in cell culture
A new model of viral infection spreading in cell cultures is proposed in the paper 8, taking into account virus mutation. This model represents a reaction-diffusion system of equations with time delay for the concentrations of uninfected cells, infected cells and viral load. Infection progression is characterized by the virus replication number Rv, which determines the total viral load. Analytical formulas for the speed of propagation and for the viral load are obtained and confirmed by numerical simulations. It is shown that virus mutation leads to the emergence of a new virus variant. Conditions of the coexistence of the two variants or competitive exclusion of one of them are found, and different stages of infection progression are identified.
5.2.3 Virus replication and competition in a cell culture: Application to the SARS-CoV-2 variants
Viral replication in a cell culture is described in the paper 9 by a delay reaction-diffusion system. It is shown that infection spreads in cell culture as a reaction-diffusion wave, for which the speed of propagation and viral load can be determined both analytically and numerically. Competition of two virus variants in the same cell culture is studied, and it is shown that the variant with larger individual wave speed outcompetes another one, and eliminates it. This approach is applied to the Delta and Omicron variants of the SARS-CoV-2 infection in the cultures of human epithelial and lung cells, allowing characterization of infectivity and virulence of each variant, and their comparison.
5.3 Modeling of cancer and its treatment
5.3.1 The effect of lenvatinib and pembrolizumab on thyroid cancer refractory to iodine 131I simulated by mathematical modeling
Immunotherapy and targeted therapy are alternative treatments to differentiated thyroid cancer (DTC), which is usually treated with surgery and radioactive iodine. However, in advanced thyroid carcinomas, molecular alterations can cause a progressive loss of iodine sensitivity, thereby making cancer resistant to radioactive iodine-refractory (RAIR). In the treatment of cancer, tyrosine kinase inhibitors are administered to prevent the growth of cancer cells. One such inhibitor, lenvatinib, forms a targeted therapy for RAIR-DTC, while the immunotherapeutic pembrolizumab, a humanized antibody, prevents the binding of programmed cell death ligand 1 (PD-L1) to the PD-1 receptor. As one of the first studies on treatments for thyroid cancer with mathematical model involving immunotherapy and targeted therapy, we developed in the paper 20, an ordinary differential system and tested variables such as concentration of lenvatinib and pembrolizumab, total cancer cells, and number of immune cells (i.e., T cells and natural killer cells). Analyzing local and global stability and the simulated action of drugs in patients with RAIR-DTC, revealed the combined effect of the targeted therapy with pembrolizumab. The scenarios obtained favor the combined therapy as the best treatment option, given its unrivaled ability to boost the immune system’s rate of eliminating tumor cells.
5.3.2 Discrete maturity and delay differential-difference model of hematopoietic cell dynamics with applications to Acute Myenogenous Leukemia
In the last few years, many efforts were oriented towards describing the hematopoiesis phenomenon in normal and pathological situations. This complex biological process is organized as a hierarchical system that begins with primitive hematopoietic stem cells (HSCs) and ends with mature blood cells: red blood cells, white blood cells and platelets. Regarding acute myelogenous leukemia (AML), a cancer of the bone marrow and blood, characterized by a rapid proliferation of immature cells, which eventually invade the bloodstream, there is a consensus about the target cells during the HSCs development which are susceptible to leukemic transformation. We propose and analyze in 2 a mathematical model of HSC dynamics taking into account two phases in the cell cycle, a resting and a proliferating one, by allowing just after division a part of HSCs to enter the resting phase and the other part to come back to the proliferating phase to divide again. The resulting mathematical model is a system of nonlinear differential-difference equations. Due to the hierarchical organization of the hematopoiesis, we consider n stages of HSCs characterized by their maturity levels. We obtain a system of 2n nonlinear differential-difference equations. We study the existence, uniqueness, positivity, boundedness and unboundedness of the solutions. We then investigate the existence of positive and axial steady states for the system, and obtain conditions for their stability. Sufficient conditions for the global asymptotic stability of the trivial steady state as well as conditions for its instability are obtained. Using neutral differential equation associated to the differential-difference system, we also obtain results on the local asymptotic stability of the positive steady state. Numerical simulations are carried out to show the influence of variations of the differentiation rates and self-renewal coefficients of the HSCs on the behavior of the system. In particular, we show that a blocking of differentiation at an early stage of HSC development can lead in an overexpression of very immature cells. Such situation corresponds to the observation in the case of AML.
5.4 Travelling waves and adaptation to variable environments
5.4.1 Traveling waves for reaction-diffusion PDE coupled to difference equation with nonlocal dispersal term and time delay
We consider in 3 a class of biological models represented by a system composed of reaction diffusion PDE coupled with difference equations (renewal equations) in n -dimensional space, with nonlocal dispersal terms and implicit time delays. The difference equation generally arises, by means of the method of characteristics, from an age-structured partial differential system. Using upper and lower solutions, we study the existence of monotonic planar traveling wave fronts connecting the extinction state to the uniform positive state. The corresponding minimum wave speed is also obtained. In addition, we investigate the effect of the parameters on this minimum wave speed and we give a detailed analysis of its asymptotic behavior.
5.4.2 Non-local competition slows down front acceleration during dispersal evolution
We investigate in 10 the super-linear spreading in a reaction-diffusion model analogous to the Fisher-KPP equation, but in which the population is heterogeneous with respect to the dispersal ability of individuals, and the saturation factor is non-local with respect to one variable. We prove that the rate of acceleration is slower than the rate of acceleration predicted by the linear problem, that is, without saturation. This hindering phenomenon is the consequence of a subtle interplay between the non-local saturation and the non-trivial dynamics of some particular curves that carry the mass at the front. A careful analysis of these trajectories allows us to identify the value of the rate of acceleration. The article is complemented with numerical simulations that illustrate some behavior of the model that is beyond our analysis.
5.4.3 Dynamics of lineages in adaptation to a gradual environmental change
We investigate in 11 a simple quantitative genetics model subjet to a gradual environmental change from the viewpoint of the phylogenies of the living individuals. We aim to understand better how the past traits of their ancestors are shaped by the adaptation to the varying environment. The individuals are characterized by a one-dimensional trait. The dynamics-births and deaths-depend on a time-changing mortality rate that shifts the optimal trait to the right at constant speed. The population size is regulated by a nonlinear non-local logistic competition term. The macroscopic behaviour can be described by a PDE that admits a unique positive stationary solution. In the stationary regime, the population can persist, but with a lag in the trait distribution due to the environmental change. For the microscopic (individual-based) stochastic process, the evolution of the lineages can be traced back using the historical process, that is, a measure-valued process on the set of continuous real functions of time. Assuming stationarity of the trait distribution, we describe the limiting distribution, in large populations, of the path of an individual drawn at random at a given time T. Freezing the non-linearity due to competition allows the use of a many-to-one identity together with Feynman-Kac's formula. This path, in reversed time, remains close to a simple Ornstein-Uhlenbeck process. It shows how the lagged bulk of the present population stems from ancestors once optimal in trait but still in the tail of the trait distribution in which they lived.
5.4.4 Evolutionary dynamics of complex traits in sexual populations in a heterogeneous environment: how normal?
When studying the dynamics of trait distribution of populations in a heterogeneous environment, classical models from quantitative genetics choose to look at its system of moments, specifically the first two ones. Additionally, in order to close the resulting system of equations, they often assume that the trait distribution is Gaussian (see for instance Ronce and Kirkpatrick 2001). The aim of the paper 13 is to introduce a mathematical framework that follows the whole trait distribution (without prior assumption) to study evolutionary dynamics of sexually reproducing populations. Specifically, it focuses on complex traits, whose inheritance can be encoded by the infinitesimal model of segregation (Fisher 1919). We show that it allows us to derive a regime in which our model gives the same dynamics as when assuming a Gaussian trait distribution. To support that, we compare the stationary problems of the system of moments derived from our model with the one given in Ronce and Kirkpatrick 2001 and show that they are equivalent under this regime and do not need to be otherwise. Moreover, under this regime of equivalence, we show that a separation bewteen ecological and evolutionary time scales arises. A fast relaxation toward monomorphism allows us to reduce the complexity of the system of moments, using a slow-fast analysis. This reduction leads us to complete, still in this regime, the analytical description of the bistable asymmetrical equilibria numerically found in Ronce and Kirkpatrick 2001. More globally, we provide explicit modelling hypotheses that allow for such local adaptation patterns to occur.
5.4.5 Analysis of a model of cell crawling migration
We introduce and study in 24 a model for motility of cells on substrate. The cell is 1d, inextensible and it contains a diffusive back-polarity marker, which satisfies a non-linear and non-local parabolic equation of Fokker-Planck type with attachment/detachment at the boundary. The idea behind the model is a quadratic nonlinear coupling: the marker is advected by the cell velocity, which is itself driven by a front-rear imbalance in marker. We show that it is of bistable type, provided that the coupling between the asymmetry of the marker and the cell velocity is sufficiently strong. In such a case we prove the non-linear stability of the largest steady state, for large initial data. In the weak coupling case we prove the convergence of the molecular concentration towards the Gaussian state.
5.5 Immuno-epidemiological models
5.5.1 Immuno-epidemiological model-based prediction of further Covid-19 epidemic outbreaks due to immunity waning
In 17, we develop a new data-driven immuno-epidemiological model with distributed infectivity, recovery and death rates determined from the epidemiological, clinical and experimental data. Immunity in the population is taken into account through the time-dependent number of vaccinated people with different numbers of doses and through the acquired immunity for recovered individuals. The model is validated with the available data. We show that for the first time from the beginning of pandemic COVID-19 some countries reached collective immunity. However, the epidemic continues because of the emergence of new variant BA.2 with a larger immunity escape or disease transmission rate than the previous BA.1 variant. Large epidemic outbreaks can be expected several months later due to immunity waning. These outbreaks can be restrained by an intensive booster vaccination.
5.5.2 An epidemic model with time delay determined by the disease duration
Immuno-epidemiological models with distributed recovery and death rates can describe the epidemic progression more precisely than conventional compartmental models. However, the required immunological data to estimate the distributed recovery and death rates are not easily available. In 18, an epidemic model with time delay is derived from the previously developed model with distributed recovery and death rates, which does not require precise immunological data. The resulting generic model describes epidemic progression using two parameters, disease transmission rate and disease duration. The disease duration is incorporated as a delay parameter. Various epidemic characteristics of the delay model, namely the basic reproduction number, the maximal number of infected, and the final size of the epidemic are derived. The estimation of disease duration is studied with the help of real data for COVID-19. The delay model gives a good approximation of the COVID-19 data and of the more detailed model with distributed parameters.
5.5.3 An epidemic model with time-distributed recovery and death rates
A compartmental epidemiological model with distributed recovery and death rates is proposed in 19. In some particular cases, the model can be reduced to the conventional SIR model. However, in general, the dynamics of epidemic progression in this model is different. Distributed recovery and death rates are evaluated from COVID-19 data. The model is validated by the epidemiological data for different countries, and it shows better agreement with the data than the SIR model. The time-dependent disease transmission rate is estimated.
5.5.4 Stability analysis of a delayed immune response model to viral infection
The purpose of the work 16 is the study of the qualitative behavior of the homogeneous in space solution of a delay differential equation arising from a model of infection dynamics. This study is mainly based on the monotone dynamical systems theory. Existence and smoothness of solutions are proved, and conditions of asymptotic stability of equilibriums in the sense of monotone dynamical systems are formulated. Then, sufficient conditions of global stability of the nonzero steady state are derived, for the two typical forms of the function f, specifying the efficiency of immune response-mediated virus elimination. Numerical simulations illustrate the analytical results. The obtained theoretical results have been applied, in a context of COVID-19 data calibration, to forecast the immunological behaviour of a real patient.
5.5.5 Global asymptotic stability for a distributed delay differential-difference system of a Kermack-McKendrick SIR model
We investigate in 4 a system of distributed delay differential-difference equations describing an epidemic model of susceptible, infected, recovered and temporary protected population dynamics. A nonlocal term (distributed delay) appears in this model to describe the temporary protection period of the susceptible individuals. We investigate mathematical properties of the model. We obtain the global asymptotic stability of the two steady states: disease-free and endemic. We construct appropriate Lyapunov functionals where the basic reproduction number appears as a threshold for the global asymptotic behavior of the solution between disease extinction and persistence.
5.5.6 Traveling waves of a differential-difference diffusive Kermack-McKendrick epidemic model with age-structured protection phase
We consider in 5 a general class of diffusive Kermack-McKendrick SIR epidemic models with an age-structured protection phase with limited duration, for example due to vaccination or drugs with temporary immunity. A saturated incidence rate is also considered which is more realistic than the bilinear rate. The characteristics method reduces the model to a coupled system of a reaction-diffusion equation and a continuous difference equation with a time-delay and a nonlocal spatial term caused by individuals moving during their protection phase. We study the existence and non-existence of non-trivial traveling wave solutions. We get almost complete information on the threshold and the minimal wave speed that describes the transition between the existence and non-existence of non-trivial traveling waves that indicate whether the epidemic can spread or not. We discuss how model parameters, such as protection rates, affect the minimal wave speed. The difficulty of our model is to combine a reaction-diffusion system with a continuous difference equation. We deal with our problem mainly by using Schauder's fixed point theorem. More precisely, we reduce the problem of the existence of non-trivial traveling wave solutions to the existence of an admissible pair of upper and lower solutions.
5.5.7 Forecasting the effect of Pre-Exposure Prophylaxis (PrEP) on HIV propagation with a system of differential–difference equations with delay
The HIV/AIDS epidemic is still active worldwide with no existing definitive cure. Based on the WHO recommendations stated in 2014, a treatment, called Pre-Exposure Prophylaxis (PrEP), has been used in the world, and more particularly in France since 2016, to prevent HIV infections. In the paper 6, we propose a new compartmental epidemiological model with a limited protection time offered by this new treatment. We describe the PrEP compartment with an age-structure hyperbolic equation and introduce a differential equation on the parameter that governs the PrEP starting process. This leads us to a nonlinear differential–difference system with discrete delay. After a local stability analysis, we prove the global behavior of the system. Finally, we illustrate the solutions with numerical simulations based on the data of the French Men who have Sex with Men (MSM) population. We show that the choice of a logistic time dynamics combined with our Hill-function-like model leads to a perfect data fit. These results enable us to forecast the evolution of the HIV epidemics in France if the populations keep using PrEP.
5.6 Why are periodic erythrocytic diseases so rare in humans?
Many studies have shown that periodic erythrocytic (red blood cell linked) diseases are extremely rare in humans. To explain this observation, we develop in 1 a simple model of erythropoiesis in mammals and investigate its stability in the parameter space. A bifurcation analysis enables us to sketch stability diagrams in the plane of key parameters. Contrary to some other mammal species such as rabbits, mice or dogs, we show that human-specific parameter values prevent periodic oscillations of red blood cells levels. In other words, human erythropoiesis seems to lie in a region of parameter space where oscillations exclusively concerning red blood cells cannot appear. Further mathematical analysis show that periodic oscillations of red blood cells levels are highly unusual and if exist, might only be due to an abnormally high erythrocytes destruction rate or to an abnormal hematopoietic stem cell commitment into the erythrocytic lineage. We also propose numerical results only for an improved version of our approach in order to give a more realistic but more complex approach of our problem.
5.7 Modelling of cytokine storm in respiratory viral infections
In the paper 23, we develop a model of the immune response to respiratory viral infections taking into account some particular properties of the SARS-CoV-2 infection. The model represents a system of ordinary differential equations for the concentrations of epithelial cells, immune cells, virus and inflammatory cytokines. Conventional analysis of the existence and stability of stationary points is completed by numerical simulations in order to study dynamics of solutions. Behavior of solutions is characterized by large peaks of virus concentration specific for acute respiratory viral infections. At the first stage, we study the innate immune response based on the protective properties of interferon secreted by virus-infected cells. On the other hand, viral infection down-regulates interferon production. Their competition can lead to the bistability of the system with different regimes of infection progression with high or low intensity. In the case of infection outbreak, the incubation period and the maximal viral load depend on the initial viral load and the parameters of the immune response. In particular, increase of the initial viral load leads to shorter incubation period and higher maximal viral load. In order to study the emergence and dynamics of cytokine storm, we consider proinflammatory cytokines produced by cells of the innate immune response. Depending on parameters of the model, the system can remain in the normal inflammatory state specific for viral infections or, due to positive feedback between inflammation and immune cells, pass to cytokine storm characterized by excessive production of proinflammatory cytokines. Furthermore, inflammatory cell death can stimulate transition to cytokine storm. However, it cannot sustain it by itself without the innate immune response. Assumptions of the model and obtained results are in qualitative agreement with the experimental and clinical data.
6 Bilateral contracts and grants with industry
6.1 Bilateral Grants with Industry
Participants: [ M. Bouvier, [ T. Espinasse, [ O. Gandrillon, [ T. Lepoutre.
- CIFRE PhD for Matteo Bouvier, with the Vidium company. Objective: Identification and control of GRNs. A first step will consist in establishing the proof of concept of a Design Of Experiment method to refine the inference of GRNs. A second step will consist in developing a method for controlling the dynamic stochastic systems that underlie GRNs behavior.
7 Partnerships and cooperations
7.1 International initiatives
Participants: [ All team members.
7.1.1 Inria associate team not involved in an IIL or an international program
- Associated team MathModelingHematopoiesis “Mathematical modeling of hematopoietic stem cell dynamics in normal and pathological hematopoiesis with optimal control for drug therapy”, 2019 - 2022 (stopped in 2020 due to COVID-19).
- Participants (Dracula): M. Adimy [Coordinator], L. Pujo-Menjouet and V. Volpert.
- Partner Institution(s) from India: Presidency University, Kolkata - Mathematics Department of Indian Institute of Technology, Roorkee - Department of Mathematics and Statistics, Indian Institute of Technology, Kanpur.
- The project proposes to develop and analyse new mathematical models of Hematopoietic Stem Cell population dynamics in normal and pathological hematopoiesis. Two important questions will be explored in this project: i) the biological data concerning the hematopoiesis process evolves constantly, and new understanding modifies the established mathematical models, ii) modeling constraints us to simplify the complicated biological scenarios, which moving away from the reality, but enabling us to reach a certain comprehension of the hematopoiesis process.
- Associated team MoCoVec “Modelling and Biological Control of Vector-Borne Diseases: the case of Malaria and Dengue”, 2022-2025.
- Participants (Dracula): M. Adimy.
- Partner Institution(s): MAMBA Inria, Paris (P.A. Bliman [Coordinator]) - FAPESP São Paulo, Brazil.
- Focusing on dengue and malaria, two diseases transmitted by vector mosquito and which cause high morbidity and mortality around the world, this project aims to model disease transmission, its spread and control, in a context of climatic and environmental change. For this, the main drives of disease transmission will be addressed to understand which factors modulate the spatio-temporal patterns observed, especially in Brazil.
7.1.2 STIC/MATH/CLIMAT AmSud projects
- STIC AmSud project NEMBICA “New methods for biological control of the arboviruses”, 2019-2022.
- Participants (Dracula): M. Adimy.
- Partner Institution(s): MAMBA Inria, Paris (P.A. Bliman [Coordinator]) - CIRAD - CNRS - Argentina - Chile - Colombia - Paraguay.
- This project is concerned with new strategies of biological control of the spread of many and important diseases transmitted by mosquitoes of the genus Aedes, which put at risk considerable portions of the human population and infect millions of people every year. The aim is to elaborate and analyze pertinent models, and to evaluate qualitatively and quantitatively different control strategies, with the goal of understanding the key aspects and parameters of the dynamics, testing concepts, estimating feasibility, identifying risks and reducing cost.
- MATH AmSud project TOMENADE “Topological methods and non autonomous dynamics for delay differential equations”, 2022-2024.
- Participants (Dracula): M. Adimy.
- Partner Institution(s): Laboratoire des Signaux et Systèmes (L2S), CNRS, Centrale Supelec - Argentina (P. Amster [Coordinator]) - Chile - Brazil.
- This project addresses open problems about non autonomous systems of delay differential equations modeling some phenomena from life sciences, namely, a meta-populations version of the Nicholson equations and models of competition in a stirred chemostat. Nevertheless, the ideas and methods could be certainly extended in several ways, and we also expect to make progress in topics as the (non autonomous) topological linearization problem and the possibility of converse results for persistence.
7.1.3 Participation in other International Programs
- ECOS-Sud France-Chilie, C20E03 “Coarsening dynamics: numerical and theoretical analysis of the Lifchitz-Slyozov equation with nucleation and applications to biology”, 2021-2023.
- Participants (Dracula): L. Pujo-Menjouet and L. Tine.
- Partner Institution(s): Inria ANACONDA - Institut Denis Poisson, University Orléans - Universidad del Bio-Bio, Concepcion, Chilie (E. Hingant, [Coordinator]).
- The general objectives of this project are i) To push-forward theoretical studies on well-posedness of solutions of the Lifshitz- Slyozov model and variants, and understand long-time behavior and scaling properties of those solutions, ii) To explore various numerical strategies to accurately simulate solutionsoft heLifshitz-Slyozov model, iii) To apply our developed methodology on biology applications, in particular within interdisciplinary work on protein aggregation in neurodegenerative diseases, and on adipocyte formation dynamics.
- IFCAM1 funds “Mathematical modeling of hematopoiesis process in application to chronic and acute myelogenous leukemia”, 2018-2021 (stopped in 2020 due to COVID-19).
- Participants (Dracula): M. Adimy [Coordinator], L. Pujo-Menjouet and V. Volpert.
- Partner Institution(s) from India: Presidency University, Kolkata - Department of Mathematics and Statistics, Indian Institute of Technology, Kanpur.
- The project proposes to develop and analyze new realistic mathematical models of blood cells formation in the bone marrow (hematopoiesis). Two important scientific questions will be explored in this project: i) the biological data concerning the hematopoiesis process evolves constantly, and new understanding modifies the established mathematical models, ii) modeling constraints us to simplify the complicated biological scenarios, which moving away from the reality, but enabling us to reach a certain comprehension of the hematopoiesis process. The challenging task is to combine biological, mathematical and numerical approaches to obtain better perceiving of the underlying mechanisms between normal and pathological hematopoiesis. Thus, we shall investigate different mathematical models, mainly based on age-structured and delay differential equations, to shed new light on the understanding of specific biological phenomena observed during pathological hematopoiesis (for instance, the appearance of oscillations in patients with Chronic Myeloid Leukemia (CML); Or, the overproduction of blasts in patients with Acute Myeloid Leukemia (AML)).
- PHC-Maghreb “Mathematical modeling and control of the spread of infectious diseases in the Mediterranean area”, 2022-2024.
- Participants (Dracula): Samuel Bernard, Vitaly Volpert, Léon Matar Tine [Coordinator].
- Partner Institution(s): Inria-Dracula, ICJ (Université Lyon 1), Mohammed V University of Rabat, Ecole Nationale d'Ingénieurs de Tunis, Université Tlemcen
- This research project aims to contribute to the development of some decision-making tools in the context of major global public health challenges: namely the modelling, monitoring and control of the spread of infectious diseases in the socio-economic current context. To do this, it is a question of contributing to: 1) improving mathematical representations of the human immune system, 2) construction of compartmental epidemiological models in which susceptible individuals are divided into several classes according to their immunity (vaccinated or not, age, infection with or without provisional immunity, infection with or without recurrence, etc.).
- Mecan'os “Si tu ne peux pas l’avoir, (re)construis le !” 2021-2023
- Participants (Dracula): L. Pujo-Menjouet [Coordinator].
- Partner Institution(s): Institut Camille Jordan - CUNY, New-York - University of Tlemcen, Algeria.
- The aim of this project is to describe the gene regulatory network involving osteoblasts and its interactions with key proteins in the mineralization of the collagen matrix. A feedback loop from the matrix stiffness induces whole control of the proteins. The question is how. The application is to be able to produce bio-materials showing any required stiffness properties for bone substitutes for instance.
- micrOS-macrOS “Modélisation multi-échelle de l'os”, 2021-2023
- Participants (Dracula): L. Pujo-Menjouet [Coordinator].
- Partner Institution(s): Institut Camille Jordan - CUNY, New-York - University of Tlemcen, Algeria.
- The objective of this project is to understand the impact of the gene regulatory network (at the molecular scale) on the bio-mechanic properties of the bone (bone scale). This new multi-scale approach will allow to finely understand the qualitative and quantitative role of osteoblasts and osteoclasts on the normal or pathological bone formation.
7.2 European initiatives
7.2.1 H2020 projects
COSMIC
COSMIC project on cordis.europa.eu
-
Title:
COmbatting disorders of adaptive immunity with Systems MedICine
-
Duration:
From January 1, 2018 to September 30, 2022
-
Partners:
- INSTITUT NATIONAL DE RECHERCHE EN INFORMATIQUE ET AUTOMATIQUE (INRIA), France
- ETHNIKO KENTRO EREVNAS KAI TECHNOLOGIKIS ANAPTYXIS (CENTRE FOR RESEARCH AND TECHNOLOGY HELLAS CERTH), Greece
- KNIME, Germany
- NBIC / DTL data projects, Netherlands
- ANAXOMICS BIOTECH SL (AX), Spain
- IBM RESEARCH GMBH (IBM), Switzerland
- REDOXIS AB (REDOXIS AB), Sweden
- Biotecture (Biotecture), Netherlands
- SURF BV, Netherlands
- THE BABRAHAM INSTITUTE, United Kingdom
- HELMHOLTZ-ZENTRUM FUR INFEKTIONSFORSCHUNG GMBH (HZI), Germany
- AMC MEDICAL RESEARCH BV (AMC Medical Research B.V.), Netherlands
- KAROLINSKA INSTITUTET (KI), Sweden
- Vivia Biotech, Spain
- ELEVATE BV (ELEVATE), Netherlands
- ACADEMISCH MEDISCH CENTRUM BIJ DE UNIVERSITEIT VAN AMSTERDAM (AMC), Netherlands
- IFOM-ISTITUTO FONDAZIONE DI ONCOLOGIA MOLECOLARE ETS (.), Italy
- Immunowell foundation, Netherlands
-
Inria contact:
Fabien CRAUSTE
- Coordinator:
-
Summary:
EU countries face large health challenges to combat chronic diseases. Recently, systems medicine has emerged as a promising discipline to accelerate the translation of basic research into applications for improved diagnostics and personalized treatment. Its power arises from the integration of laboratory and computational approaches crossing research disciplines and sectors to solve clinical questions. COSMIC delivers the next generation of leading, entrepreneurial, and innovative systems medicine professionals having expertise, skills, and experience to successfully combat complex human disorders. These professional will find excellent career opportunities. COSMIC focuses on B-cell neoplasia and rheumatoid arthritis, prototypical diseases originating from abnormal functioning of immune cells, often resulting in similar antigen specificities. COSMIC enables Early Stage Researchers to play a leading role in this exciting field. COSMIC develops and integrate experimental and computational approaches and establish a unique cross-fertilization between oncology and auto-immunity. In addition to transferable skills, the training program focuses on establishing a double expertise in laboratory and computational to address clinical questions. It involves a wide-range of stakeholders: (pre)clinical departments, companies, patient groups, students, and the general public. COSMIC will establish a link with the leading European EASyM and ISBE initiatives, and aims to harmonize systems medicine training throughout Europe by connecting to other EU (Marie Curie systems medicine) training initiatives. COSMIC (i) significantly improves ESR career perspectives (ii) leads to new public-private collaborations increasing competitiveness for companies; (iii) contributes to future oncology and immunology medical care; (iv) contributes to the EU systems medicine best practices.
7.3 National initiatives
7.3.1 ANR
ANR Prio Diff
-
Title:
Impact of replication and structural diversification of prions on their cerebral dissemination
-
Partner Institution(s):
- Inrae Jouy-en-Josas, France
- Inrae Toulouse, France
- CEA, Fontenay aux Roses, France
- Institut Camille Jordan, France
-
Date/Duration:
July 2021-June 2025
-
Additionnal info/keywords:
Prions are lethal proteinaceous pathogens with major public-health risks due to their zoonotic and iatrogenic potential. They are composed of aggregated, misfolded conformers of the host-encoded prion protein that progressively deposit in the brain by a self-perpetuating reaction. The underlying molecular mechanisms of replication and tissue dissemination remain mostly elusive. Our objective is to model these processes entirely based on recent advances that prion aggregates are conformationally heterogeneous and dynamic rather than uniform and static. To achieve this, we will map in prion-infected brain the structural diversification-to-bioactivity/neurotoxicity landscape of prion assemblies in a spatiotemporal manner and mathematically build a multiscale model of diversification and lesion spreading. The goal is to generate an open access model capable of predicting the disease progression and identify key elementary process for therapeutics intervention and early diagnostics. / Keywords: interactions hôtes pathogènes, infectiologie, modélisation prion, tissue diffusion, neurotropism.
ANR SingleStatOmics
-
Title:
Statistics and Machine Learning for Single Cell Genomics
-
Partner Institution(s):
- University Montpellier
- University Dauphine
- Ecole Central de Nantes
- Google Brain / Mines ParisTech
- AgroParisTech / INRAe
- University Grenoble
- University Lyon 1 (F. Picard [coordinator])
- Ecole Centrale de Lyon.
-
Date/Duration:
2019-2023
-
Additionnal info/keywords:
The goal is to develop new methodologies to investigate cell identity and the dynamics of cell differentiation, by integrating single cell expression and epigenomic data.
ANR Memoires
-
Title:
Multiscale Modeling of CD8 T cell immune responses
-
Partner Institution(s):
- ICL-CIRI (J. Marvel [Coordinator])
- Ecole Centrale de Lyon
- AltraBio.
-
Date/Duration:
2019-2023
-
Additionnal info/keywords:
The main goal will is to identify, at the single cell level, signatures associated with functionally relevant memory cell subsets, to characterize dynamical molecular networks that are predictive of their generation, and to build a multi-scale computational model of this process.
ANR PLUME
-
Title:
Molecular and morphogenetic control of feather pattern formation
-
Partner Institution(s):
- Centre interdisciplinaire de recherche en biologie (M. Manceau [Coordinator])
- Institut de Biologie du Développement de Marseille
- Brandeis University.
-
Date/Duration:
2021-2025
-
Additionnal info/keywords:
The objectives are to identify the hierarchy of pattern forming mechanisms establishing initial patterning spaces, characterise material properties and mechanical stresses controlling primordia self-organisation, and uncover how how cell and mechanical dynamics control the timely wave of primordia production. This work will shed light on the mechanisms governing pattern variation and fidelity in nature.
7.3.2 Other projects
Digitbio
-
Title:
MP DIGIT-BIO, AMI 2021
-
Partner Institution(s):
- Inrae Jouy-en-Josas, France
- Inria, Dracula team, France
-
Date/Duration:
September 2021-August 2023
-
Additionnal info/keywords:
Ce projet a pour but de permettre d’explorer pour la première fois dans le champ des maladies neurodégénératives comment la dynamique des assemblages de PrPSc régit la dissémination, la ségrégation et l’accumulation de certaines sous-populations d’assemblages de PrPSc et conduit à l’apparition d’une structure souche-spécifique ainsi que de possibles fronts de propagation / Interactions hôtes pathogènes, infectiologie, modélisation prion, tissue diffusion, neurotropism.
7.4 Regional initiatives
- T. Lepoutre [Coordinator] and O. Gandrillon have been granted by IXXI on “Relaxation experiments for gene expression”, 2021.
-
-This project aims at modelling relaxation experiments, performed on a cell line which is a good model for Chronic Myeloid Leukemia (CML) cells. We will model these experiments via EDP models structured in gene expression in order to use the information from the expression histograms during the experiment.
-
-
8 Dissemination
8.1 Promoting scientific activities
8.1.1 Scientific events: organisation
Member of the organizing committees
- Laurent Pujo-Menjouet is the organizer of the Workshop Bonitos, May 20, Institut Camille Jordan, Villeurbanne, France.
- IHP thematic trimester Mathematical modeling of organization in living matter (January 10th to April 1st, 2022).
- V. Calvez is part of the organizing comittee and specially involved in the organization of the conference Mathematical models in ecology and evolution (March 21st to 25th).
- T. Lepoutre has been part of the organizing comittee of one of the conferences Tissue growth and movement (Jan. 10th to 14th)
8.1.2 Journal
Member of the editorial boards
- Vitaly Volpert is Editor-in-chief:
-
-Mathematical Modelling of Natural Phenomena (MMNP).
-
-
- Mostafa Adimy is a member of the Editorial Boards:
-
-Journal of Nonlinear Systems and Applications (JNSA),
-
-Mathematical Modelling of Natural Phenomena (MMNP),
-
-Pure and Applied Mathematics Journal.
-
-
- Laurent Pujo-Menjouet is a member of the Editorial Boards:
-
-Journal for theoretical Biology,
-
-PLOS One,
-
-MDPI,
-
-Mathematical Modelling of Natural Phenomena.
-
-
- Olivier Gandrillon is a member of the Editorial Boards of
-
-BMC research Notes.
-
-
8.2 Teaching - Supervision - Juries
8.2.1 Supervision
- PhD of Kyriaki Dariva, "Contributions à l'étude de la stabilité dans des modèles de populations de cellules". University Lyon 1, until June 20, 2022, supervisor: Thomas Lepoutre.
- PhD of Léonard Dekens, "Adaptation d'une population avec une reproduction sexuée à un environnement hétérogène : modélisation mathématique et analyse asymptotique dans un régime de petite variance", University Lyon 1, until July 07, 2022, supervisor: Vincent Calvez.
- PhD of Mete Demircigil, "Etude du mouvement collectif chez Dictyostelium discoideum et autres espèces. Modélisation, Analyse et Simulation", ENS-Lyon, until August 26, 2022, supervisor: Vincent Calvez.
- PhD of Elias Ventre, "Analyse, calibration et évaluation de modèles stochastiques d'expression des gènes", ENS-Lyon, until September 28, 2022, supervisors: Olivier Gandrillons, Thibault Espinasse and Thomas Lepoutre.
- PhD in progress: Cheikh Gueye, "Problèmes inverses pour l'estimation de paramètres de modèles mathématiques: Application en dynamique des populations", University Lyon 1 and University Cheikh Anta Diop of Dakar, since October 16, 2018, supervisors: Ionel-sorinel Ciuperca, Abdoulaye Sene and de Léon matar Tine.
- PhD in progress: Matteo Bouvier, "Identification et contrôle de réseaux de régulation de gènes", CIFRE with the Vidium company and ENS-Lyon, since October 01, 2020, supervisor: Olivier Gandrillon.
- PhD in progress: Margaux Prieux, "Etude des réseaux de régulation de la différenciation des CD8 mémoire par une approche moléculaire au niveau cellule unique", ENS-Lyon, since October 01, 2019, supervisors: Olivier Gandrillon and Jacqueline Marvel.
- PhD in progress: Grégoire Ranson, "Mathematical modeling of epidemics spreading dealing with temporary treatment effciency", University Lyon 1 and York University, since April 27, 2022, supervisors: Mostafa Adimy, Laurent Pujo-Menjoeut and Jianhong Wu.
- PhD in Progress: Charlotte Dugourd, "Nested Immuno-epidemiological modeling of the dynamics of intra- and inter-host infections", Université Lyon 1, since October 01, 2022, supervisor: Mostafa Adimy.
- PhD in progress: Arsène Marzorati, University Lyon 1, since October 01, 2022, supervisors: Samuel Bernard and Jonathan Rouzaud-Cornabas.
- PhD in progress: Basile Fornara, "Mécanismes de dissémination des prions dans les tissus cérébraux : une approche synthétique",Université Paris Cité, since October 21, 2022, supervisors: Human Rezaei et de Laurent Pujo-menjouet.
- PhD in progress: Maxime Estavoyer, "Modélisation mathématique de la formation des plumes d'oiseaux: interaction entre propagation instruction et auto-organisation", University Lyon 1, since March 03, 2022, supervisor: Thomas Lepoutre.
- Post-doc: Thao Nguyen Thi Nhu, since February 2022, supervisors: Olivier Gandrillon.
- Post-doc: Nicolas Torres, since April 2022, supervisors: Laurent Pujo-Menjouet.
- Post-doc: Michèle Romanos, since December 2022, supervisors: Vincent Calvez.
9 Scientific production
9.1 Publications of the year
International journals
Doctoral dissertations and habilitation theses
Reports & preprints
9.2 Cited publications
- 35 articleStability Analysis of a Model of Interaction Between the Immune System and Cancer Cells in Chronic Myelogenous Leukemia.Bulletin of Mathematical Biology2017, 1--27
- 36 articleLong-term treatment effects in chronic myeloid leukemia.Journal of Mathematical Biology2017, 1-26
- 37 articleImplication of the Autologous Immune System in BCR-ABL Transcript Variations in Chronic Myelogenous Leukemia Patients Treated with Imatinib..Cancer Research7519October 2015, 4053-62
- 38 articleIdentification of Nascent Memory CD8 T Cells and Modeling of Their Ontogeny.Cell Systems43February 2017, 306-317
- 39 articleInferring gene regulatory networks from single-cell data: a mechanistic approach.BMC Systems Biology111November 2017, 105