
Keywords
Computer Science and Digital Science
- A2.1.1. Semantics of programming languages
- A2.1.5. Constraint programming
- A2.1.10. Domain-specific languages
- A2.2.1. Static analysis
- A2.3.2. Cyber-physical systems
- A2.4. Formal method for verification, reliability, certification
- A2.4.1. Analysis
- A2.4.2. Model-checking
- A2.4.3. Proofs
- A3.4.2. Unsupervised learning
- A3.4.4. Optimization and learning
- A6.1.1. Continuous Modeling (PDE, ODE)
- A6.1.2. Stochastic Modeling
- A6.1.3. Discrete Modeling (multi-agent, people centered)
- A6.1.4. Multiscale modeling
- A6.2.4. Statistical methods
- A6.2.6. Optimization
- A6.3.1. Inverse problems
- A6.3.4. Model reduction
- A7.2. Logic in Computer Science
- A8.1. Discrete mathematics, combinatorics
- A8.2. Optimization
- A8.7. Graph theory
- A9.7. AI algorithmics
Other Research Topics and Application Domains
- B1. Life sciences
- B1.1.2. Molecular and cellular biology
- B1.1.7. Bioinformatics
- B1.1.8. Mathematical biology
- B1.1.10. Systems and synthetic biology
- B2.2.3. Cancer
- B2.2.6. Neurodegenerative diseases
- B2.4.1. Pharmaco kinetics and dynamics
- B9. Society and Knowledge
1 Team members, visitors, external collaborators
Research Scientists
- François Fages [Team leader, INRIA, Senior Researcher, HDR]
- Aurélien Naldi [INRIA, Starting Research Position, until Sep 2022]
- Sylvain Soliman [INRIA, Researcher, HDR]
Faculty Members
- Anna Niarakis [UNIV EVRY, Associate Professor, HDR]
- Denis Thieffry [ENS PARIS, HDR]
PhD Students
- Sahar Aghakhani [UNIV EVRY]
- Marine Collery [IBM]
- Jeremy Grignard [Servier, CIFRE, until Sep 2022]
- Julien Martinelli [INSERM, until Mar 2022]
- Eléa Thibault-Greugny [Johnson&Johnson, CIFRE, until Oct 2022]
Technical Staff
- Mathieu Hemery [INRIA, Engineer]
Interns and Apprentices
- Julien Bienvenu [INRIA, Ecole Polytechnique, Intern, from Apr 2022 until Aug 2022]
Administrative Assistant
- Natalia Alves [INRIA]
2 Overall objectives
This project aims at developing formal methods for understanding the cell machinery and establishing computational paradigms in cell biology. It is based on the vision of
cells as machines,
chemical reaction networks as programs,
and on the use of concepts from computer science to master the complexity of cell biochemical processes 23.
We contribute to the development of a computational theory of chemical reaction networks (CRNs), by addressing fundamental research issues in computer science on the concepts of analog computation and analog computational complexity in biochemistry, and on the interplay between structure and dynamics in CRNs.
Since 2002, we develop a software platform, called the Biochemical Abstract Machine (BIOCHAM), for modeling, analyzing and now synthesizing CRNs, with some unique algorithmic contributions. The reaction rule-based language of BIOCHAM allows us to reason about CRNs at different levels of abstraction in the hierarchy of their stochastic, differential, Boolean and hybrid semantics. Various static analysis methods, most of them based on constraint solving or graph theory, provide useful information before going to simulations and dynamical analyses, for which quantitative temporal logic is used to formalize cell behaviors with imprecise data, and to constrain model building.
A tight integration between dry lab and wet lab efforts is also essential for the success of the project. This is achieved through collaborations with biologists and experimentalists, including partners from the pharmaceutical industry, on concrete biological and biomedical questions.
Because of the importance of constraint solving and optimization techniques in our approach, and the need of rapid prototyping for our software developments, we keep some research and teaching activity in relational programming (i.e. logic programming, constraint programming, interval arithmetic) as a general paradigm for computing with partial information systems, and solving practical instances of NP-hard problems.
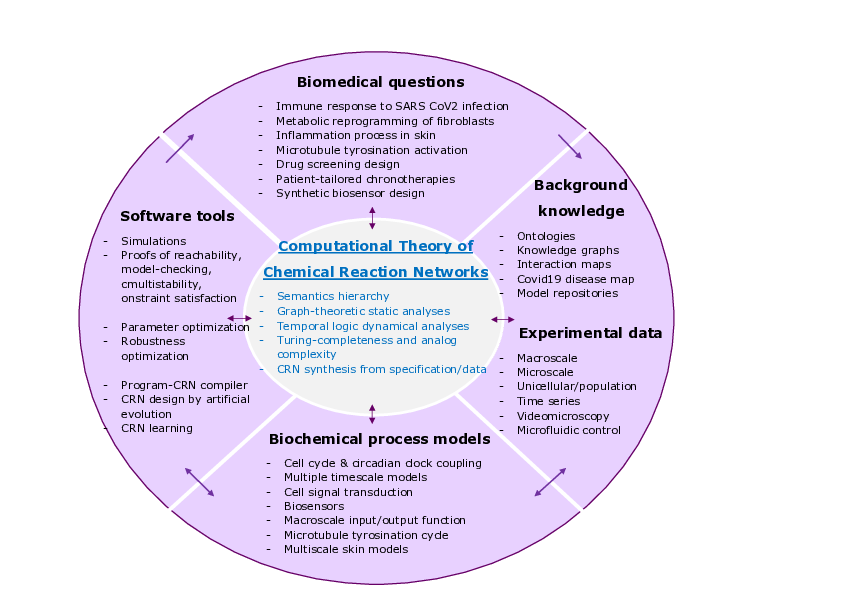
Wheel of biomedical questions, background knowledge and experimental data, biological process models and decision making tools, with the computational theory of chemical reaction networks at the center.
3 Research program
3.1 Chemical Reaction Network (CRN) Theory
Originally, Feinberg's mathematical theory of Chemical Reaction Networks (CRN) and Thomas's influence networks, were created to provide sufficient and/or necessary structural conditions for the existence of multiple steady states and oscillations in complex gene networks. Those conditions can be verified by static analyzers without knowing kinetic parameter values, nor making any simulation. In this first approach, most of our work consists in considering the hypergraph structure of a CRN (Petri net invariants, species-reaction labelled hypergraph, influence graph, reductions by subgraph epimorphisms) and analysing their interplay with the dynamics of CRNs in their different interpretations (Boolean, CTMC, ODE, time scale separations) which can be related in the framework of abstract interpretation1. For example, our study of the influence graphs of reaction systems 6 lead recently to sufficient graphical conditions ensuring rate-independence of CRN 3, or some time ago to the non-trivial generalization to reaction systems of Thomas' conditions of multi-stationarity and Soulé's proof given for influence systems, with much greater efficiency by several orders of magnitude for testing them when compared to current symbolic computation methods 1.
However, we aim at development a computational theory of CRNs and biochemical programming. Our original method to infer CRNs from ODEs 4 showed the generality of CRNs, and lead us to prove the Turing-completeness of continuous CRNs over a finite set of molecular species, showing that any computable function over the real numbers (i.e. computable in arbitrary precision by a Turing machine) can be computed by a CRN over a finite set of molecular species 5. (Best paper award CMSB 2017, Prix La Recherche 2019). This result closed the last open problem on the computation power of CRNs in their different semantics and has opened a whole research avenue on CRN design by compilation of mathematical functions. This is illustrated by a series of publications since that date, again this year 20 (best paper award CMSB 2022), and innovative applications in synthetic biology 2.
3.2 Logical Paradigm for Systems Biology
Our group was among the first ones in 2002 to apply model-checking methods to systems biology in order to reason on large molecular interaction networks, such as Kohn's map of the mammalian cell cycle (800 reactions over 500 molecules) 2. The logical paradigm for systems biology that we have subsequently developed for quantitative models can be summarized by the following identifications :
biological model = transition system , initial state
dynamical behavior specification = temporal logic formula
model validation = model-checking
model reduction = sub-model-checking, s.t.
model prediction = formula enumeration, s.t.
static experiment design = symbolic model-checking,
model synthesis = constraint solving
model inference = constraint solving
In particular, the definition of a continuous satisfaction degree for first-order temporal logic formulae with constraints over the reals, was the key to generalize this approach to quantitative models, opening up the field of model-checking to model optimization 34
This line of research continues with the development of temporal logic constraint patterns with efficient solvers, and their use for model building, in partnership with biologists to answer concrete questions in the biomedical domain5610, 9 and the pharmaceutical industry 13, 18, 21.
3.3 Constraint Solving and Optimization
Constraint solving and optimization algorithms are important in our research. On the one hand, static analyses of CRNs often involve solving hard combinatorial optimization problems, for which we have shown that constraint logic programming techniques are particularly successful, often beating dedicated algorithms on real-size instances from model repositories by orders of magnitude 77.
On the other hand, parameter search problems involve solving hard continuous optimization problems, for which evolutionary algorithms, and especially the covariance matrix evolution strategy (CMA-ES) (EPI RANDOPT) have shown to provide best results in our context. Constraint-based models and efficient constraint solvers are thus instrumental in our approach for building quantitative models, gaining model-based insights, revisiting biological hypotheses, and contributing to biological knowledge.
4 Application domains
4.1 Preamble
Our collaborative work on biological applications is expected to serve as a basis for groundbreaking advances in cell functioning understanding, cell monitoring and control, and novel therapy design and optimization. Our collaborations with biologists are focused on concrete biological questions, and on the building of mechanistic models of biological systems to answer them. Furthermore, one important application of our research is the development and distribution of a modeling software for computational systems biology and synthetic biology.
4.2 Modeling software for systems and synthetic biology at the cellular level
Since 2002, we develop an open-source software environment for modeling and analyzing biochemical reaction systems. This software, called the Biochemical Abstract Machine (BIOCHAM), is compatible with SBML for importing and exporting models from repositories such as BioModels. It can perform a variety of static analyses, specify behaviors in Boolean or quantitative temporal logics, search parameter values satisfying temporal constraints, and make various simulations. While the primary reason of this development effort is to be able to implement our ideas and experiment them quickly on a large scale, using rapid prototyping techniques based on constraint logic programming, BIOCHAM is distributed and used by other groups worldwide, for building CRN models, for comparing CRN analysis/synthesis techniques, and for teaching computational systems biology. A Jupyter BIOCHAM kernel has been developed to use BIOCHAM on our web server without any installation which is heavily used for teaching. We plan to continue developing BIOCHAM for these different purposes with the recruitment of a research engineer to improve the software quality and animation of the community of users.
Since 2018, the CaSQ software complements this effort by providing an interface to import large interaction maps written in SBML using the CellDesigner tool and translate them into Boolean influence models with various tools compatible with the SBML-qual standard.
Since 2020, we participate in the CoLoMoTo notebook platform, which provides an integrated collection of software tools for the analysis of qualitative models, including CaSQ. This platform encourages the reproducibility of analysis by combining Docker images (reproducible software environment) and Jupyter notebooks (reproducible and shareable workflows).
These two last efforts play a central role in the global Covid-19 Disease Maps project, see Section 8.6.
4.3 Biomedical applications
We plan to continue to tackle challenging concrete biomedical questions with academic and industrial partners in an opportunist way, according to both the scope of the question on the international scene, and the relevance of our theoretical approaches to the question.
As mentioned above, our long-standing collaboration with F. Molina's CNRS-ALCEN lab on the design, optimization and industrialization of biochemical diagnosis vesicles 2 continues on a design methodology of robust CRNs including analog functions (submitted project ANR BIOPSYN).
Our successful collaboration with Servier on knowledge graphs and CRN model parameterizations using quantitative temporal logic for drug screening 13 is planned to continue with a second CIFRE thesis with Servier on CRN model learning from experimental data.
Our similarly successful collaboration with Johnson& Johnson Santé Beauté France on multi-scale modeling of the epidermis and multifactorial aspects of atopic dermatitis 18, 21 has revealed complex metastability behaviors in our population dynamics models and further emerging properties when combined with a multi-agent model at the tissue level. That research continues with O. Radulescu in Montpellier on the tropical algebraic analysis of metastability in our model.
We also keep ready to continue our long-standing collaborations on chronotherapies as specialists of coupled models of the cell cycle and the circadian clock 26, 10 and their systemic regulators8.
Last but not least, our group became very active in cooperative efforts at the European scale to design large molecular interaction maps for diseases such as Covid-19 8 or rheumatoid arthritis 19 and derive from them Boolean dynamical models using the CaSQ software as well as developing hybrid models 12.
5 Social and environmental responsibility
5.1 Footprint of research activities
In synthetic biology, our approach based on analog computation target enzymatic reactions with proteins in artificial vesicles. They are thus DNA-free and RNA-free, solving thus major safety issue be them for biomdecial applications or the environment.
5.2 Impact of research results
Our multidisciplinary research rooted in fundamental computer science aims at contributing to biology and medicine by going quite far in the applications with partners from academia and industry, in particular pharma industry.
5.3 Carbon footprint
Our team actively selected train journeys for the participation in conferences and seminars whenever possible.
6 Highlights of the year
6.1 Algebraic Biochemistry: a Framework for Analog Online Computation in Cells (Best Paper Award CMSB 2022).
Our article 20 received the Best Paper Award of the 20th Int. Conf. on Computational Methods in Systems Biology, CMSB 2022. The Turing completeness of continuous chemical reaction networks (CRNs) states that any computable real function can be computed by a continuous CRN on a finite set of molecular species, possibly restricted to elementary reactions, i.e. with at most two reactants and mass action law kinetics. In 20, we introduce a notion of online analog computation for the CRNs that stabilize the concentration of their output species to the result of some function of the concentration values of their input species, whatever changes are operated on the inputs during the computation. We prove that the set of real functions stabilized by a CRN with mass action law kinetics is precisely the set of real algebraic functions.
This result provides an unexpectedly simple mathematical characterization of the real functions that can be computed online in a robust analog manner. This is of direct relevance to CRN design problems for innovative biosensor and biomedicine applications.
6.2 Stability versus Meta-stability in a Skin Microbiome Model
The skin microbiome plays an important role in the maintenance of a healthy skin. It is an ecosystem, composed of several species, competing for resources and interacting with the skin cells. Imbalance in the cutaneous microbiome, also called dysbiosis, has been correlated with several skin conditions, including acne and atopic dermatitis. Generally, dysbiosis is linked to colonization of the skin by a population of opportunistic pathogenic bacteria (for example C. acnes in acne or S. aureus in atopic dermatitis). Treatments consisting in non-specific elimination of cutaneous microflora have shown conflicting results. It is therefore necessary to understand the factors influencing shifts of the skin microbiome composition.
In 21, we introduce a mathematical model based on ordinary differential equations, with 2 types of bacteria populations (skin commensals and opportunistic pathogens) to study the mechanisms driving the dominance of one population over the other. By using published experimental data, assumed to correspond to the observation of stable states in our model, we derive constraints that allow us to reduce the number of parameters of the model from 13 to 5. Interestingly, a meta-stable state settled at around 2 days following the introduction of bacteria in the model, is followed by a reversed stable state after 300 hours. On the time scale of the experiments, we show that certain changes of the environment, like the elevation of skin surface pH, create favorable conditions for the emergence and colonization of the skin by the opportunistic pathogen population. Such predictions help identifying potential therapeutic targets for the treatment of skin conditions involving dysbiosis of the microbiome, and question the importance of meta-stable states in mathematical models of biological processes.
The mathematical analysis of this interesting meta-stability phenomenon in our population dynamics model is currently under investigation by tropical algebra methods with colleagues from our former ANR-DFG Symbiont project.
6.3 Hybrid Model of Rheumatoid Arthritis published in PLOS Computational Biology
Rheumatoid Arthritis (RA) is an autoimmune disease characterized by a highly invasive pannus formation consisting mainly of Synovial Fibroblasts (RASFs). RASFs’ activation is associated with metabolic alterations resulting from dysregulation of extracellular signals’ transduction and gene regulation. Deciphering the intricate mechanisms at the origin of this metabolic reprogramming may provide sign insight into RASFs’ involvement in RA’s pathogenesis and offer new therapeutic strategies. Qualitative and quantitative dynamic modeling can address some of these features, but hybrid models represent a real asset in their ability to span multiple layers of biological machinery.
In 12 we present the first hybrid RASF model: the combination of a cell-specific qualitative regulatory network with a global metabolic network. The automated framework for hybrid modeling exploits the regulatory network’s trap-spaces as additional constraints on the metabolic network. Subsequent flux balance analysis allows assessment of RASFs’ regulatory outcomes’ impact on their metabolic flux distribution. The hybrid RASF model reproduces the experimentally observed metabolic reprogramming induced by signaling and gene regulation in RASFs. Simulations also enable further hypotheses on the potential reverse Warburg effect in RA. RASFs may undergo metabolic reprogramming to turn into "metabolic factories", producing high levels of energy-rich fuels and nutrients for neighboring demanding cells through the crucial role of HIF1.
In 19 we also update the global RA map to a state-of-the-art RA-Atlas that covers relevant metabolic pathways and cell-specific molecular interaction maps for CD4+ Th1 cells, fibroblasts, and M1 and M2 macrophages. The RA-Atlas is freely accessible on the webserver MINERVA allowing easy navigation using semantic zoom, cell-specific or experimental data overlay, gene set enrichment analysis, pathway export or drug query.
6.4 Mathematical model of the microtubule tyrosination cycle for cell-based drug screening design, published in PLOS Computational Biology with Servier
Microtubules and their post-translational modifications are involved in major cellular processes. In severe diseases such as neurodegenerative disorders, tyrosinated tubulin and tyrosinated microtubules are in lower concentration. In 13 we present a mechanistic mathematical model of the microtubule tyrosination cycle combining computational modeling and high-content image analyses to understand the key kinetic parameters governing the tyrosination status in different cellular models. That mathematical model is parameterized, firstly, for neuronal cells using kinetic values taken from the literature, and, secondly, for proliferative cells, by a change of two parameter values obtained, and shown minimal, by a continuous optimization procedure based on temporal logic constraints to formalize experimental high-content imaging data. In both cases, the mathematical models explain the inability to increase the tyrosination status of microtubules by activating the Tubulin Tyrosine Ligase enzyme. The tyrosinated tubulin is indeed the product of a chain of two reactions in the cycle: the detyrosinated microtubule depolymerization followed by its tyrosination. The tyrosination status at equilibrium is thus limited by both reaction rates and activating the tyrosination reaction alone is not effective. Our computational model also predicts the effect of inhibiting the Tubulin Carboxy Peptidase enzyme which we have experimentally validated in MEF cellular model. Furthermore, the model predicts that the activation of two particular kinetic parameters, the tyrosination and detyrosinated microtubule depolymerization rate constants, in synergy, should suffice to enable an increase of the tyrosination status in living cells.
7 New software and platforms
7.1 New software
7.1.1 BIOCHAM
-
Name:
The Biochemical Abstract Machine
-
Keywords:
Bioinformatics, Systems Biology, Computational biology
-
Functional Description:
The Biochemical Abstract Machine (BIOCHAM) is a software environment for modeling, analyzing and synthesizing biochemical reaction networks (CRNs) with respect to a formal specification of the observed or desired behavior of a biochemical system. BIOCHAM is compatible with the Systems Biology Markup Language (SBML) and contains some unique features about formal specifications in quantitative temporal logic, sensitivity and robustness analyses and parameter search in high dimension w.r.t. behavioral specifications, static analyses, and synthesis of CRNs.
-
Release Contributions:
– compilation pipeline of elementary mathematical functions in at most bi-molecular reaction networks over a finite set of abstract molecular species – improved heuristic quadratization algorithm to generate at most bi-molecular reactions – notebooks of Master classes in Bioinfomratics of molecular interaction networks – multiple improvements of the commands and documentation
- URL:
-
Contact:
François Fages
-
Participants:
François Fages, Mathieu Hemery, Sylvain Soliman
7.1.2 CaSQ
-
Name:
CellDesigner as SBML-Qual
-
Keywords:
SBML, Logical Framework, Knowledge representation
-
Functional Description:
CaSQ is a tool that can convert a molecular interaction map built with CellDesigner, or any similar SBML-capable tool, to an executable Boolean model. CaSQ is developed in Python (download and install instructions can be found on the Python package index) and uses as source the xml file of CellDesigner, in order to infer preliminary Boolean rules based solely on network topology and semantic annotations (e.g., certain arcs are noted as catalysis, inhibition, etc.). The aim is to convert a Process Description representation, i.e., a reaction model, into a full logical model. The resulting structure is closer to an Activity Flow diagram, though not in a strict SBGN-PD to SBGN-AF notion. Moreover logical rules that make the model executable are also obtained. CaSQ was used on maps of the Rheumatoïd Arthritis, of the MAP-Kinase cascade, etc. and is now being used by the Covid-19 DiseaseMaps consortium to automatically obtain logical models from maps [2].
CaSQ has recently been added to the CoLoMoTo Docker image and can be used in such a notebook.
- URL:
- Publications:
-
Contact:
Sylvain Soliman
-
Participants:
Sylvain Soliman, Anna Niarakis, Aurélien Naldi
-
Partner:
Université d'Evry-Val d'Essonne
7.1.3 CoLoMoTo Notebook
-
Keywords:
Systems Biology, Computational biology
-
Functional Description:
Dynamical models are widely used to summarize our understanding of biological networks, to challenge its consistency, and to guide the generation of realistic hypothesis before experimental validation. Qualitative models in particular enable the study of the dynamical properties that do not depend on kinetic parameters, which can be difficult to obtain. This platform integrates a wide collection of complementary software tools, along with interoperability features and a consistent Programming interface based on the popular Python language.
The platform is distributed as a Docker image, ensuring the reproducibility of the software environment. The use of Jupyter Notebooks provides a good balance between ease of use and flexibility. These notebooks can be further used to share precise analysis workflows, ensuring the dissemination of fully reproducible research work using this platform.
- URL:
- Publication:
-
Contact:
Aurélien Naldi
-
Participants:
Sylvain Soliman, Aurélien Naldi, Denis Thieffry
-
Partners:
LaBRI, Institut Curie, ENS Paris, LRI - Laboratoire de Recherche en Informatique
8 New results
8.1 Algebraic Biochemistry: a Framework for Analog Online Computation in Cells.
Participants: Julien Bienvenu, François Fages, Mathieu Hemery, Sylvain Soliman.
As mentionned earlier, the Turing completeness of continuous chemical reaction networks (CRNs) states that any computable real function can be computed by a continuous CRN on a finite set of molecular species, possibly restricted to elementary reactions, i.e. with at most two reactants and mass action law kinetics 5.
In 20, we introduce a notion of online analog computation for the CRNs that stabilize the concentration of their output species to the result of some function of the concentration values of their input species, whatever changes are operated on the inputs during the computation. We prove that the set of real functions stabilized by a CRN with mass action law kinetics is precisely the set of real algebraic functions.
This result provides an unexpectedly simple mathematical characterization of those functions of high interest for CRN design problems. However, whether the computation error can be controlled in the same way is an open problem on which we are working.
8.2 Boolean model analysis, computation of complex attractors
Participants: Aurélien Naldi, Sylvain Soliman.
Boolean modelling of gene regulation but also of post-transcriptomic systems has proven over the years that it can bring powerful analyses and corresponding insight to the many cases where precise biological data is not sufficiently available to build a detailed quantitative model. This is even more true for very large models where such data is frequently missing and led to a constant increase in size of logical models à la Thomas. Besides simulation, the analysis of such models is mostly based on attractor computation, since those correspond roughly to observable biological phenotypes. The recent use of trap spaces made a real breakthrough in that field allowing to consider medium-sized models that used to be out of reach. However, with the continuing increase in model-size, the state-of-the-art computation of minimal trap spaces based on prime-implicants shows its limits as there can be a huge number of implicants.
In 22 we present an alternative method to compute minimal trap spaces, and hence complex attractors, of a Boolean model. It replaces the need for prime-implicants by a completely different technique, namely the enumeration of maximal siphons in the Petri net encoding of the original model. We demonstrated the efficiency of this approach and of its implementation using Answer Set Programming by comparing it to implicant-based methods on some large Boolean models from the literature.
In 31 we present another approach based on the notion of linear cut. The interaction graph of a Boolean network admits a linear cut when a linear component occurs in each cycle and in each path from components with multiple targets to components with multiple regulators. Under this structural condition the attractors are in one-to-one correspondence with the minimal trap spaces, and the reachability of attractors can also be easily characterized. Linear cuts provide the basis for a new interpretation of the Boolean semantics that captures all behaviours of multi-valued refinements with regulatory thresholds defined for each interaction.
8.3 On learning mechanistic models from time series data
Participants: Marine Collery, François Fages, Jeremy Grignard, Julien Martinelli, Sylvain Soliman.
Mathematical modeling of biological processes aims at providing formal representations of complex systems to enable their study, both in a qualitative and quantitative fashion. The need for explainability suggests the recourse to mechanistic models, which explicitly describe molecular interactions. Nevertheless, such models currently rely on the existence of prior knowledge on the underlying reaction network structure. Moreover, their conception remains an art which necessitates creativity combined to multiple interactions with analysis and data fitting tools. This rules out numerous applications conceivable in personalized medicine, and calls for methodological advances towards machine learning of patient-tailored models.
In his thesis 26 in collaboration with INSERM Institut Curie, Julien Martinelli shows how algorithms can be devised to learn models of dynamical interactions from temporal data, with an emphasis on explainability for the human modeler. Its applications are in the context of personalized chronotherapies, that consist in optimizing drug administration with respect to the patient's biological rhythms over the 24-hour span. Three main themes are explored: mechanistic modeling, network inference and treatment personalization. The first chapter describes the development of the first quantitative mechanistic model of the cellular circadian clock integrating transcriptomic, proteomic and sub-cellular localization data. This model has been successfully connected to a model of cellular pharmacology of an anticancerous drug, irinotecan, achieving personalization of its optimal administration timing. The second chapter introduces a novel protocol for inferring whole-body systemic controls enforced on peripheral clocks. On the long run, this approach will make it possible to integrate individual data collected from wearables for personalized chronotherapies. The third chapter presents a general algorithm to infer reactions with chemical kinetics from time series data 30.
In the ongoing thesis of Marine Collery in collaboration with IBM France, we investigate a different approach based on neural networks to learn explainable rules from sequential data in different contexts 28.
8.4 On bridging scales in modeling multifactorial aspects of atopic dermatitis
Participants: François Fages, Eléa Greugny, Mathieu Hemery.
Atopic dermatitis (AD) is a common chronic disease affecting up to 20% of young children, depending on countries, and around 10% of adults globally. It manifests as dry, itchy and sometimes cracked skin lesions on the face and body. Despite efficient immunotherapy treatments for the most severe forms of the disease, the complex mechanisms involved are still incompletely understood and improvements can be made regarding management strategies for milder forms. AD involves diverse interacting factors such as impaired skin barrier function, exacerbated inflammation, and microbiome dysbiosis, making it challenging to develop adequate in vitro models. A computational approach allows the aggregation of current pathophysiology knowledge, facilitates the visualization of the phenomena involved, and enables the prediction of certain behaviors of the system under specific conditions.
In her Doctorate thesis 24 Eléa Thibault-Greugny first introduces an agent-based model of the epidermis, able to represent aspects of atopic skin such as skin barrier dysfunction and microbiome dysbiosis. By integrating enzymatic reactions contributing to the desquamation process, into an existing agent-based model of the epidermis at the cellular level, we can study the impact of skin surface pH on the epidermal structure and function 18. The model predicts that an elevation of skin surface pH above physiologic levels accelerates the desquamation process through its action on serine proteases. This results in a significant reduction of the skin’s capacity to retain water, and increases its permeability to external penetration, including irritants. This skin barrier impairment further leads to a more intense inflammatory reaction under conditions of high skin surface pH, compared to physiologic pH levels.
Next, we introduce a mathematical model of the microbiome, based on ordinary differential equations, with 2 types of bacteria populations (skin commensals and opportunistic pathogens) to study the mechanisms driving the dominance of one population over the other. On the time scale of the experiments, the model predicts that certain changes of the environment, like the elevation of skin surface pH from physiologic levels, create favorable conditions for the emergence and colonization of the skin by opportunistic pathogens. Interestingly, for certain parameter values, a meta-stable state settled at around 2 days following the introduction of bacteria in the model, is followed by a reversed stable state after 300 hours 21. The mathematical analysis of this phenomenon is currently under investigation using tropical algebra and constraint solving methods 29 developed in the framework of the ANR-DFG SYMBIONT project.
Finally, we integrate the mathematical model of the microbiome into the agent-based model. Special consideration is taken regarding the timescales of the processes described and their location in the epidermis. The resulting model is used to study how the constant surface cells renewal impacts the microbiome kinetics. This thesis provides additional evidence that skin surface pH, serine proteases and skin microbiome could be interesting therapeutic targets for AD maintenance therapy.
8.5 Computational modeling of the tyrosination cycle of microtubules for drug discovery
Participants: François Fages, Jeremy Grignard.
The drug discovery process is long, costly and very risky. In his thesis 25 Jeremy Grignard shows how the relevance of the primary phases of drug discovery can be improved by developing new computational methods. The first contribution concerns the development of the Pegasus knowledge graph in order to capitalize on the heterogeneous and multi-sourced pharmaco-biological data of the pharmaceutical sector. The industrial applications of Pegasus address the problems of therapeutic projects and allow to characterize off-target effects of perturbators, to design a new experiment, and to identify focused screening libraries. The second contribution concerns the development of an algorithm for the identification of positive control compounds and a normalization algorithm to improve the design and analysis of high content phenotypic screening experiments. These algorithms allow the normalization of phenotypic signatures obtained from screening campaigns and the integration of informative phenotypic similarities in the Pegasus knowledge graph. The third contribution 13 concerns the development of a mathematical model of the microtubule tyrosination cycle which explains, on the one hand, the inactivity of chemical compounds shown to be active outside the cell, and on the other hand, suggests the need to activate two reactions of this cycle, in synergy, to obtain an effect in cellular models. This illustrates the contribution of mathematical modeling to, on the one hand, predict and understand the counter-intuitive dynamics of biochemical processes that are not representable by static knowledge graphs as in Pegasus, and on the other hand, guide the design of new screening experiments. The scientific contributions and industrial applications of this thesis are developed in the context of the primary phases of drug discovery and are intended to extend to the clinical phases of the pharmaceutical process.
8.6 An interoperable computational framework for the analysis and modeling of disease mechanisms
Participants: Sahar Aghakhani, Anna Niarakis, Sylvain Soliman.
In 32 we present an update on the COVID-19 Disease Map project. This is a large-scale community effort uniting 277 scientists from 130 Institutions around the globe. We use high-quality, mechanistic content describing SARS-CoV-2-host interactions and develop interoperable bioinformatic pipelines for novel target identification and drug repurposing. Community-driven and highly interdisciplinary, the project is collaborative and supports community standards, open access, and the FAIR data principles. The coordination of community work allowed for an impressive step forward in building interfaces between Systems Biology tools and platforms. Our framework links key molecules highlighted from broad omics data analysis and computational modeling to dysregulated pathways in a cell-, tissue- or patient-specific manner. We also employ text mining and AI-assisted analysis to identify potential drugs and drug targets and use topological analysis to reveal interesting structural features of the map. The proposed framework is versatile and expandable, offering a significant upgrade in the arsenal used to understand virus-host interactions and other complex pathologies.
The use of maps and models from so many different groups led us to present in 15 recent efforts toward a common framework for annotated, accessible, reproducible and interoperable computational models in biology, and discuss key challenges of the field.
8.7 Methological aspects of population dynamics modeling
Participants: Aurélien Naldi.
In 17 we present UPMaBoSS, a framework dedicated to modeling dynamic populations of interacting cells. We rely on the preexisting tool MaBoSS, which enables probabilistic simulations of cellular networks. A novel software layer is added to account for cell interactions and population dynamics, but without considering the spatial dimension. This modeling approach can be seen as an intermediate step towards more complex spatial descriptions
Computational models have great potential to accelerate bioscience, bioengineering, and medicine. However, it remains challenging to reproduce and reuse simulations, in part, because the numerous formats and methods for simulating various subsystems and scales remain siloed by different software tools. For example, each tool must be executed through a distinct interface. In 16 we developed BioSimulators, a central registry of the capabilities of simulation tools and consistent Python, command-line and containerized interfaces to each version of each tool. The foundation of BioSimulators is standards, such as CellML, SBML, SED-ML and the COMBINE archive format, and validation tools for simulation projects and simulation tools that ensure these standards are used consistently. To help modelers find tools for particular projects, we have also used the registry to develop recommendation services. We anticipate that BioSimulators will help modelers exchange, reproduce, and combine simulations.
8.8 Methodological aspects of computational modeling in systems medicine
Participants: Sahar Aghakhani, Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
Digital twins, customized simulation models pioneered in industry, are beginning to be deployed in medicine and healthcare, with some major successes, for instance in cardiovascular diagnostics and in insulin pump control. Personalized computational models are also assisting in applications ranging from drug development to treatment optimization. More advanced medical digital twins will be essential to making precision medicine a reality. Because the immune system plays an important role in such a wide range of diseases and health conditions, from fighting pathogens to autoimmune disorders, digital twins of the immune system will have an especially high impact. However, their development presents major challenges, stemming from the inherent complexity of the immune system and the difficulty of measuring many aspects of a patient’s immune state in vivo.
In 14 we outline a roadmap for meeting these challenges and building a prototype of an immune digital twin. It is structured as a four-stage process that proceeds from a specification of a concrete use case to model constructions, personalization, and continued improvement.
In 11 we review Boolean models of disease mechanisms and compare a range of methods and tools used for analysis processes. We explain the methodology of Boolean analysis focusing on its application in disease modelling. Finally, we discuss its practical application in analysing signal transduction and gene regulatory pathways in health and disease.
In 15 we present recent efforts toward a common framework for annotated, accessible, reproducible and interoperable computational models in biology, and discuss key challenges of the field.
9 Bilateral contracts and grants with industry
9.1 Bilateral contracts with industry
9.1.1 Institut de Recherches Servier
Participants: François Fages, Jeremy Grignard, Mathieu Hemery, Julien Martinelli, Sylvain Soliman.
Cifre PhD thesis of Jeremy Grignard with Servier 25.
9.1.2 Johnson&Johnson Santé Beauté France
Participants: François Fages, Elea Greugny, Mathieu Hemery, Sylvain Soliman.
Cifre PhD thesis of Eléa Thibault-Greugny with Johnson&Johnson Santé Beauté France 24.
9.1.3 IBM research, France
Participants: Marine Collery, François Fages, Sylvain Soliman.
On-going PhD thesis of Marine Collery at IBM France on rule learning from sequential data.
10 Partnerships and cooperations
10.1 International initiatives
10.1.1 Participation in other International Programs
ANR-DFG SYMBIONT
Participants: François Fages, Mathieu Hemery, Sylvain Soliman.
- ANR-DFG Symbiont (2018-2022) on “Symbolic Methods for Biological Systems”, coordinated by T. Sturm (CNRS, LORIA, Nancy, France) and A. Weber (Univ. Bonn, Germany) with F. Fages (Inria), F. Boulier (U. Lille), O. Radulescu (U. Montpellier), A. Schuppert (RWTH Aachen), S. Walcher (RWTH Aachen), W. Seiler (U. Kassel);
10.2 European initiatives
Participants: Sahar Aghakhani, Aurélien Naldi, Anna Niaraki, Sylvain Soliman.
10.3 National initiatives
10.3.1 ANR Projects
Participants: François Fages, Mathieu Hemery, Sylvain Soliman.
- ANR ifference on "Complexity theory with discrete ODEs", coordinated by Olivier Bournez, Ecole Polytechnique, with F. Fages (Inria), F. Chyzak (Inria), A. Durand Univ. Paris-Diderot, F. Madelaine P. Valarché Univ. Créteil, N20 Jerôme Durand-Lose, Orléans - Moulay Barkatou Thomas Cluzeau, Limoges - Mathieu Sablik, Toulouse
11 Dissemination
11.1 Promoting scientific activities
Participants: François Fages, Mathieu Hemery, Aurélien Naldi, Anna Niaraki, Sylvain Soliman.
11.1.1 Scientific events: organisation
Anna Niarakis was a co-organizer of
- ECCB 2022 Workshop, Sitges
- ISMB COSI SysMod Workshop, Madison, US, July 2022
- ICSB Academy webinar series hosting Dr Benjamin Gyori, Harvard Medical School, January 2022
Member of the conference program committees
François Fages was PC member of Anna Niarakis was a member of the programme committee of JOBIM 2022
Sylvain Soliman was PC member of BIOSTEC BIOINFORMATICS 2022
11.1.2 Journal
Member of the editorial boards
François Fages is member of the Editorial Board of the Computer Science area of the Royal Society Open Science journal, since 2014.
Anna Niarakis is member of the Editorial Board of Scientific Reports Nature Publishing and Immunoinformatics, Elsevier.
Reviewer - reviewing activities
She wrote reviews for Molecular Medicine, Plos Computational Biology, Immunoinformatics, Frontiers in Applied Mathematics and Statistics, Nucleic Acid Research and Cancers.
François Fages wrote reviews for JACM, Natural Computing, PLOS computational biology.
Mathieu Hemery wrote reviews for Physical Review E (2 reviews), Physical Review R (2 reviews), RSOS (1 review) and Natural Computing (1 review).
Sylvain Soliman wrote reviews for JOMB and PLoS Computational Biology
11.1.3 Invited talks
• Anna Niarakis, Highlight talk, JOBIM 2022, Rennes, Inference of an Integrative, Executable Network for Rheumatoid Arthritis Combining Data-Driven Machine Learning Approaches and a State-of-the-Art Mechanistic Disease Map, 5-8 July 2022 .
• GT BIOSS yearly meeting, "From mechanisms to systems to digital twins. Αddressing critical barriers in systems modelling", November 17th 2022, Nantes .
• ECMT Heidelberg Mini Symposium Digital twins in medicine: where we are, where we are heading, and what is needed "Development of a virtual Rheumatoid Arthritis synovial fibroblast for large-scale dynamic analysis and efficient drug-target identification", 19-23 September, Heidelberg .
Sylvain Soliman, GT BIOSS yearly meeting, "Logical model inference with CaSQ", November 17th 2022, Nantes.
11.1.4 Leadership within the scientific community
François Fages is member of the Steering Committee of the conference series Computational Methods for Systems Biology (CMSB) since 2008.
Anna Niarakis is:
- Member of the Scientific Advisory Board of REACTOME
- Co-leader of the Covid 19 Disease map project
- Co-leader of the Consortium Disease Maps
- Coordinator of SysMod (Systems Modelling)
- Member of the Conseil de perfectionnement Mention Bioinformatique Université Paris-Saclay
- President of the jury for the Licence Double Diplome Sciences de La Vie - Informatique Université Paris Saclay
11.1.5 Scientific expertise
François Fages was member of the jury for the recruitment of an Associate Professor in Computer Science at the Univ. Evry Val d'Essonne.
Anna Niarakis evaluated for Parcoursup Licence Double Diplome Paris Saclay SDV-Informatique
11.1.6 Research administration
François Fages is member of
- "Bureau du Comité des équipes-projets du centre Inria Saclay"
- "Bureau du département Informatique Données et Intelligence Artificielle (IDIA) de l'Institut Polytechnique de Paris"
- Ecole Doctorale ED IPP 626 ED, Institut Polytechnique de Paris, since its creation in 2019
Aurelien Naldi and Anna Niarakis are members of CoLoMoTo (Consortium for Logical Models and Tools).
Anna Niarakis is member of the French Bioinformatics Society.
Sylvain Soliman is member of the “Commission Scientifique” of Inria Saclay .
11.2 Teaching - Supervision - Juries
Participants: François Fages, Mathieu Hemery, Aurélien Naldi, Anna Niarakis, Sylvain Soliman.
11.2.1 Teaching
- International advanced course: Anna Niarakis, Ben Hall (co-organizers), Sahar Aghakhani (helper), Sylvain Soliman (trainer) Computational Systems Biology for Complex Human Disease, MRC University of Cambridge, Wellcome Trust course WGC Advanced Course, 04-09 December 2022, Hinxton, Cambridge UK.
- International advanced course: Anna Niarakis (speaker) Logical modelling of (inter) cellular networks for biotechnology and personalised medicine, August 15-26 2022, NTNU, Trondheim, Norway.
- Master 2: François Fages (coordinator 24h and teacher 12h), Jérôme Feret (12h) C2-19 - Biochemical Programming, Master Parisien de Recherche en Informatique (MPRI), Paris.
- Cycle ingénieur Ecole Polytechnique, Master 1: François Fages (coordinator, 18h lectures, 14h TD), Mathieu Hemery (6h TD) INF555 - Constraint-based Modeling and Algorithms for Decision Making ProblemsMaster Artificial Intelligence, Master Science and Technology, Ecole Polytechnique.
- Bachelor 3: François Fages (co-coordinator, 12h lectures), Mathieu Hemery (12h TD), CSE 307 - Relational Programming, Ecole Polytechnique.
- Anna Niarakis is the pedagogical responsible for the Double Bachelor in Life Sciences and Informatics at Paris Saclay University.
11.2.2 Supervision
- PhD in progress: Sahar Aghakhani, “Fibroblasts as therapeutic targets in rheumatoid arthritis (RA) and cancer. Computational modeling of the metabolic reprogramming (glycolytic switch) in RA synovial fibroblasts (RASFs) and cancer associated fibroblasts (CAFs)” , ED Paris-Saclay, since Sep. 2020, Anna Niarakis and Sylvain Soliman (50%)
- PhD in progress : Marine Collery, “Apprentissage de règles à partir de données dépendantes du temps appliqué à la détection de fraudes”, ED IPP, Ecole Polytechnique, since Sept. 2020, P. Bonnard, F. Fages (1/3) and R. Kuiters, IBM research France
- PhD E. Thibault-Greugny: ‘Computational modeling approaches to multifactorial aspects of atopic dermatitis’. Institut Polytechnique de Paris, 4th Oct. 2022. F. Fages (1/3), J. Bensaci, G. Stamatas, Johnson&Johnson Santé Beauté France
- PhD J. Grignard: ‘Computational methods to improve the early drug discovery’. Institut Polytechnique de Paris, 17th June 2022., ED IPP, Ecole Polytechnique, F. Fages (50%) and T. Dorval, Servier.
- PhD J. Martinelli: ‘On learning mechanistic models from time series data with applications to personalised chronotherapies’. Institut Polytechnique de Paris, 18th Feb. 2022., ED IPP, Ecole Polytechnique, F. Fages (50%) and A. Ballesta, INSERM.
Anna Niarakis was a mentor for the 2022 iGem Evry -Paris Saclay team (gold medal).
François Fages, Jeremy Grignard and Sylvain Soliman supervised 5 students from Ecole Polytechnique on a long term Projet Scientifique Collectif (PSC) on "Apprentissage automatique de réseaux d’interactions en pharmacologie" with Servier.
François Fages and Mathieu Hemery supervised 6 students from Ecole Polytechnique on a PSC on "Compiling Mathematical Functions in Biochemical Reaction Networks".
11.2.3 Juries
François Fages participated in the juries of:
- HDR Nadjib Lazaar, Université de Montpellier, Rapporteur, 18 novembre 2022;
- PhD Paul Dufossé, Ecole Polytechnique, Président, 22 décembre 2022.
- PhD Déborah Boyenval, Univ. Côte d'Azur, Rapporteur&Président, 15 décembre 2022.
- PhD Thibault L'Yvonnet, Univ. Côte d'Azur, 28 mars 2022.
- PhD Jérémy Pardo, Univ. Evry Val d'Essonne, 3 février 2022.
Aurélien Naldi participated in the jury of:
- PhD Maximilien Cosme, Univ. Montpellier, 28 mars 2022.
Anna Niarakis participated in the thesis commitee of:
- PhD Monraz Gomez Luis Cristobal, Institut Curie, CDV - Complexité du vivant n°515, 24 March 2022
11.3 Popularization
11.3.1 Articles and contents
Our book chaper 23 is intended to provide a gentle introduction to a completely new topic: analog biochemical programming, making for the first time of programming a natural science...
11.3.2 Education
Mathieu Hemery took part to the Inria stand for the "Fête de la Science" on the campus of the École polytechnique. And do two presentations for the "semaine des math" in the Collège Moreau at Monthléry (91) presenting simple theorems for secondary school students and one presentation in the Lycée Marie Laurencin at Mennecy (91) for the Chiche programme in which scientific talk of their work and daily activity to high school students.
12 Scientific production
12.1 Major publications
- 1 articleGraphical Requirements for Multistationarity in Reaction Networks and their Verification in BioModels.Journal of Theoretical Biology459December 2018, 79--89
- 2 articleComputer‐aided biochemical programming of synthetic microreactors as diagnostic devices.Molecular Systems Biology144April 2018
- 3 inproceedingsGraphical Conditions for Rate Independence in Chemical Reaction Networks.Proceedings CMSB 2020: The 18th International Conference on Computational Methods in Systems BiologyCMSB 2020 - 18th International Conference on Computational Methods in Systems BiologyKonstanz / Online, GermanySeptember 2020
- 4 articleInferring reaction systems from ordinary differential equations.Theoretical Computer Science599September 2015, 64--78
- 5 inproceedingsStrong Turing Completeness of Continuous Chemical Reaction Networks and Compilation of Mixed Analog-Digital Programs.CMSB 2017 - 15th International Conference on Computational Methods in Systems BiologyLecture Notes in Computer ScienceProceedings of the fiveteen international conference on Computational Methods in Systems Biology, CMSB 201710545Darmstadt, GermanySeptember 2017, 108-127
- 6 articleInfluence Networks compared with Reaction Networks: Semantics, Expressivity and Attractors.IEEE/ACM Transactions on Computational Biology and BioinformaticsPP992018, 1-14
- 7 articleOn Enumerating Minimal Siphons in Petri nets using CLP and SAT solvers: Theoretical and Practical Complexity.Constraints212April 2016, 251--276
- 8 articleCOVID19 Disease Map, a computational knowledge repository of virus-host interaction mechanisms.Molecular Systems Biology1710October 2021, e10387
- 9 articleLogical model specification aided by model- checking techniques: application to the mammalian cell cycle regulation.Bioinformatics32172016, i772-i780
- 10 articleModel-based investigation of the circadian clock and cell cycle coupling in mouse embryonic fibroblasts: Prediction of RevErb-α up-regulation during mitosis.BioSystems149November 2016, 59--69
12.2 Publications of the year
International journals
International peer-reviewed conferences
Scientific book chapters
Doctoral dissertations and habilitation theses
Reports & preprints