Section: Research Program
Analysis of integrated metabolic and gene regulatory networks
Participants : Eugenio Cinquemani, Hidde de Jong, Johannes Geiselmann, Stéphan Lacour, Yves Markowicz, Manon Morin, Michel Page, Corinne Pinel, Stéphane Pinhal, Delphine Ropers [Correspondent] , Valentin Zulkower.
The response of bacteria to changes in their environment involves responses on several different levels, from the redistribution of metabolic fluxes and the adjustment of metabolic pools to changes in gene expression. In order to fully understand the mechanisms driving the adaptive response of bacteria, as mentioned above, we need to analyze the interactions between metabolism and gene expression. While often studied in isolation, gene regulatory networks and metabolic networks are closely intertwined. Genes code for enzymes which control metabolic fluxes, while the accumulation or depletion of metabolites may affect the activity of transcription factors and thus the expression of enzyme-encoding genes.
The fundamental principles underlying the interactions between gene expressions and metabolism are far from being understood today. From a biological point of view, the problem is quite challenging, as metabolism and gene expression are dynamic processes evolving on different time-scales and governed by different types of kinetics. Moreover, gene expression and metabolism are measured by different experimental methods generating heterogeneous, and often noisy and incomplete data sets. From a modeling point of view, difficult methodological problems concerned with the reduction and calibration of complex nonlinear models need to be addressed.
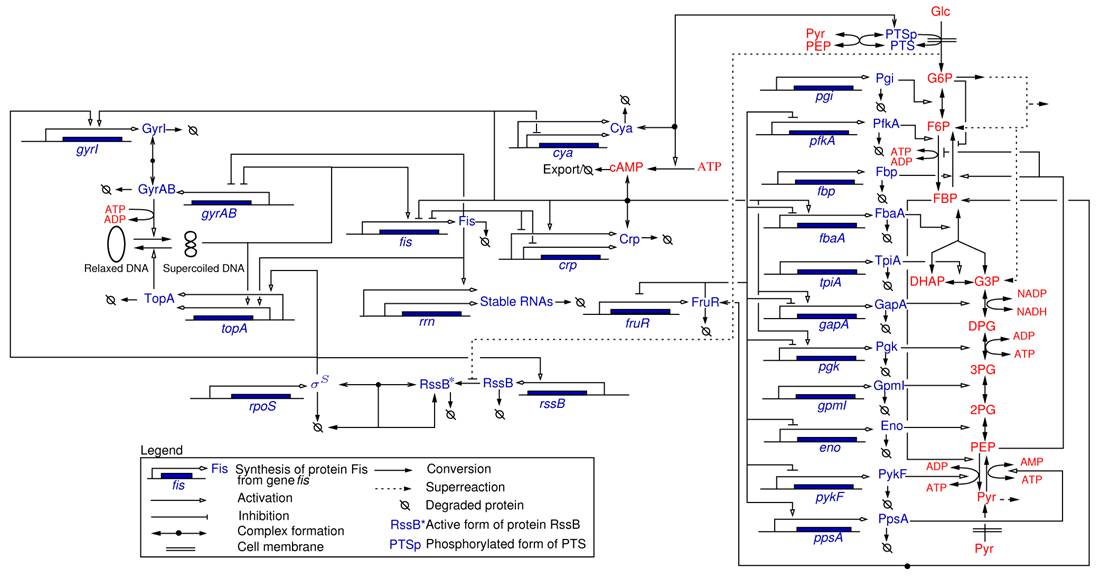
Most of the work carried out within the IBIS project-team specifically addressed the analysis of integrated metabolic and gene regulatory networks in the context of E. coli carbon metabolism (Figure 4 ). While an enormous amount of data has accumulated on this model system, the complexity of the regulatory mechanisms and the difficulty to precisely control experimental conditions during growth transitions leave many essential questions open, such as the physiological role and the relative importance of mechanisms on different levels of regulation (transcription factors, metabolic effectors, global physiological parameters, ). We are interested in the elaboration of novel biological concepts and accompanying mathematical methods to grasp the nature of the interactions between metabolism and gene expression, and thus better understand the overall functioning of the system. Moreover, we have worked on the development of methods for solving what is probably the hardest problem when quantifying the interactions between metabolism and gene expression: the estimation of parameters from hetereogeneous and noisy high-throughput data. These problems are tackled in collaboration with experimental groups at Inra/INSA Toulouse and CEA Grenoble, which have complementary experimental competences (proteomics, metabolomics) and biological expertise.
|