

Section: Research Program
Modeling sequences with formal grammars
Once groups of genome products implied in the answer of the species have been identified with integrative or dynamics methods, it remains to characterize the biological actors within genomes. To that goal, we both learn, model and parse formal patterns within DNA, RNA or protein sequences. More precisely, our research on modeling biomolecular sequences with expressive formal grammars focuses on learning such grammars from examples, helping biologists to design their own grammar and providing practical parsing tools.
On the development of machine learning algorithms for the induction of grammatical models [40], we have a strong expertise on learning finite state automata. We have proposed an algorithm that learns successfully automata modeling families of (non homologous) functional families of proteins [4], leading to a tool named Protomata-learner (see Fig. 3). The algorthim is based on a similar fragment merging heuristic approach which reports partial and local alignments contained in a family of sequences.. As an example, this tool allowed us to properly model the TNF protein family, a difficult task for classical probabilistic-based approaches. It was also applied successfully to model important enzymatic families of proteins in cyanobacteria [3]. Our future goal is to further demonstrate the relevance of formal language modeling by addressing the question of a fully automatic prediction from the sequence of all the enzymatic families, aiming at improving even more the sensitivity and specificity of the models. As enzyme-substrate interactions are very specific central relations for integrated genome/metabolome studies and are characterized by faint signatures, we shall rely on models for active sites involved in cellular regulation or catalysis mechanisms. This requires to build models gathering both structural and sequence information in order to describe (potentially nested or crossing) long-term dependencies such as contacts of amino-acids that are far in the sequence but close in the 3D protein folding. Our current researches is focused on the inference of Context-Free Grammars including the topological information coming from the structural characterization of active sites.
|
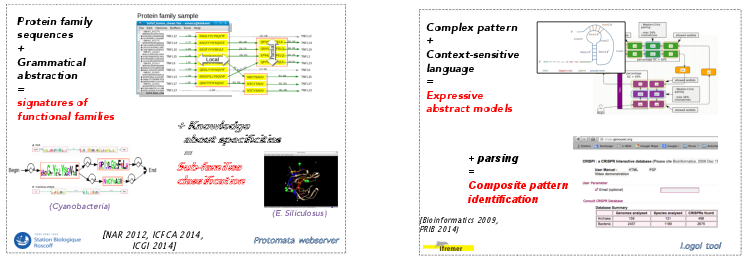
Using context-free grammars instead of regular patterns increases the complexity of parsing issues. Indeed, efficient parsing tools have been developed to identify patterns within genomes but most of them are restricted to simple regular patterns. Definite Clause Grammars (DCG), a particular form of logical context-free grammars have been used in various works to model DNA sequence features [76]. An extended formalism, String Variable Grammars (SVGs), introduces variables that can be associated to a string during a pattern search (see Fig. 3) [90], [89]. This increases the expressivity of the formalism towards mildly context sensitive grammars. Thus, those grammars model not only DNA/RNA sequence features but also structural features such as repeats, palindromes, stem/loop or pseudo-knots. Few years ago, we have designed a first tool, STAN (suffix-tree analyser), in order to make it possible to search for a subset of SVG patterns in full chromosome sequences [8]. This tool was used for the recognition of transposable elements in Arabidopsis thaliana [92]. We have enlarged this experience through a new modeling language, called Logol [1]. Generally, a suitable language for the search of particular components in languages has to meet several needs : expressing existing structures in a compact way, using existing databases of motifs, helping the description of interacting components. In other words, the difficulty is to find a good tradeoff between expressivity and complexity to allow the specification of realistic models at genome scale. The Logol language and associated framework have been built in this direction. See Figure 3 for illustration. The Logol specificity beside other SVG-like languages mainly lies in a systematic introduction of constraints on string variables.