Section: New Results
Machine learning and pluri-disciplinary research
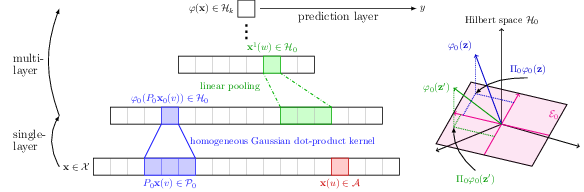
Predicting Transcription Factor Binding Sites with Convolutional Kernel Networks
Participants : Dexiong Chen, Laurent Jacob, Julien Mairal.
The growing amount of biological sequences available makes it possible to learn genotype-phenotype relationships from data with increasingly high accuracy. By exploiting large sets of sequences with known phenotypes, machine learning methods can be used to build functions that predict the phenotype of new, unannotated sequences. In particular, deep neural networks have recently obtained good performances on such prediction tasks, but are notoriously difficult to analyze or interpret. In this work, we introduce a hybrid approach between kernel methods and convolutional neural networks for sequences, which retains the ability of neural networks to learn good representations for a learning problem at hand, while defining a well characterized Hilbert space to describe prediction functions. Our method (see Figure 20), dubbed CKN-seq, outperforms state-of-the-art convolutional neural networks on a transcription factor binding prediction task while being much faster to train and yielding more stable and interpretable results.
Source code is freely available at https://gitlab.inria.fr/dchen/CKN-seq.
Loter: Inferring local ancestry for a wide range of species
Participants : Thomas Dias-Alves, Julien Mairal, Michael Blum [CNRS] .
Admixture between populations provides opportunity to study biological adaptation and phenotypic variation. Admixture studies can rely on local ancestry inference for admixed individuals, which consists of computing at each locus the number of copies that originate from ancestral source populations. Existing software packages for local ancestry inference are tuned to provide accurate results on human data and recent admixture events. Here, we introduce Loter, an open-source software package that does not require any biological parameter besides haplotype data in order to make local ancestry inference available for a wide range of species. Using simulations, we compare the performance of Loter to HAPMIX, LAMP-LD, and RFMix. HAPMIX is the only software severely impacted by imperfect haplotype reconstruction. Loter is the less impacted software by increasing admixture time when considering simulated and admixed human genotypes. LAMP-LD and RFMIX are the most accurate method when admixture took place 20 generations ago or less; Loter accuracy is comparable or better than RFMix accuracy when admixture took place of 50 or more generations; and its accuracy is the largest when admixture is more ancient than 150 generations. For simulations of admixed Populus genotypes, Loter and LAMP-LD are robust to increasing admixture times by contrast to RFMix. When comparing length of reconstructed and true ancestry tracts, Loter and LAMP-LD provide results whose accuracy is again more robust than RFMix to increasing admixture times. We apply Loter to admixed Populus individuals and lengths of ancestry tracts indicate that admixture took place around 100 generations ago.
The Loter software package and its source code are available at https://github.com/bcm-uga/Loter.
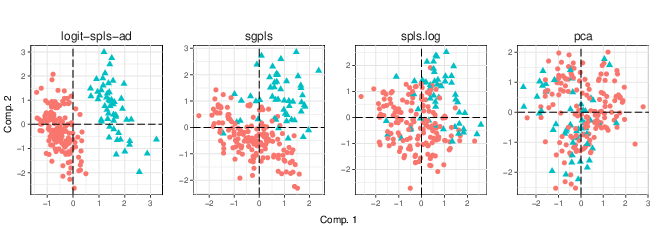
High Dimensional Classification with combined Adaptive Sparse PLS and Logistic Regression
Participant : Ghislain Durif.
The high dimensionality of genomic data calls for the development of specific classification methodologies, especially to prevent over-optimistic predictions. This challenge can be tackled by compression and variable selection, which combined constitute a powerful framework for classification, as well as data visualization and interpretation. However, current proposed combinations lead to unstable and non convergent methods due to inappropriate computational frameworks. We hereby propose a computationally stable and convergent approach for classification in high dimensional based on sparse Partial Least Squares (sparse PLS). In this work [6], we start by proposing a new solution for the sparse PLS problem that is based on proximal operators for the case of univariate responses. Then we develop an adaptive version of the sparse PLS for classification, called logit-SPLS, which combines iterative optimization of logistic regression and sparse PLS to ensure computational convergence and stability. Our results are confirmed on synthetic and experimental data. In particular we show how crucial convergence and stability can be when cross-validation is involved for calibration purposes. Using gene expression data we explore the prediction of breast cancer relapse (c.f. figure 21 for an example of data visualization). We also propose a multi-categorical version of our method, used to predict cell-types based on single-cell expression data. Our approach is implemented in the plsgenomics R-package.
|
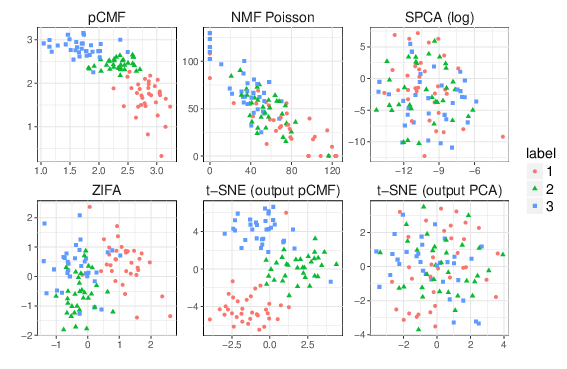
Probabilistic Count Matrix Factorization for Single Cell Expression Data Analysis
Participant : Ghislain Durif.
The development of high-throughput biology technologies now allows the investigation of the genome-wide diversity of transcription in single cells. This diversity has shown two faces: the expression dynamics (gene to gene variability) can be quantified more accurately, thanks to the measurement of lowly-expressed genes. Second, the cell-to-cell variability is high, with a low proportion of cells expressing the same gene at the same time/level. Those emerging patterns appear to be very challenging from the statistical point of view, especially to represent and to provide a summarized view of single-cell expression data. PCA is one of the most powerful framework to provide a suitable representation of high dimensional datasets, by searching for latent directions catching the most variability in the data. Unfortunately, classical PCA is based on Euclidean distances and projections that work poorly in presence of over-dispersed counts that show drop-out events (zero-inflation) like single-cell expression data. In this work [32], we propose a probabilistic Count Matrix Factorization (pCMF) approach for single-cell expression data analysis, that relies on a sparse Gamma-Poisson factor model. This hierarchical model is inferred using a variational EM algorithm. We show how this probabilistic framework induces a geometry that is suitable for single-cell data visualization, and produces a compression of the data that is very powerful for clustering purposes. Our method is competed to other standard representation methods like t-SNE, and we illustrate its performance for the representation of zero-inflated over-dispersed count data (c.f. figure 22). We also illustrate our work with results on a publicly available data set, being single-cell expression profile of neural stem cells. Our work is implemented in the pCMF R-package.
|