Section: New Results
Pluri-disciplinary Research
Biological Sequence Modeling with Convolutional Kernel Networks
Participants : Dexiong Chen, Laurent Jacob [CNRS, LBBE Laboratory] , Julien Mairal.
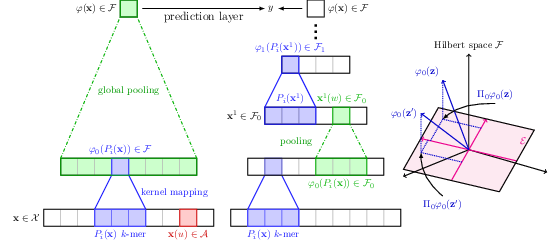
The growing number of annotated biological sequences available makes it possible to learn genotype-phenotype relationships from data with increasingly high accuracy. When large quan- tities of labeled samples are available for training a model, convolutional neural networks can be used to predict the phenotype of unannotated sequences with good accuracy. Unfortunately, their performance with medium- or small-scale datasets is mitigated, which requires inventing new data-efficient approaches. In [40], we introduce a hybrid approach between convolutional neural networks and kernel methods to model biological sequences. Our method 22 enjoys the ability of convolutional neural networks to learn data representations that are adapted to a specific task, while the kernel point of view yields algorithms that perform significantly better when the amount of training data is small. We illustrate these advantages for transcription factor binding prediction and protein homology detection, and we demonstrate that our model is also simple to interpret, which is crucial for discovering predictive motifs in sequences. The source code is freely available at https://gitlab.inria.fr/dchen/CKN-seq.
|
Token-level and sequence-level loss smoothing for RNN language models
Participants : Maha Elbayad, Laurent Besacier [LIG] , Jakob Verbeek.
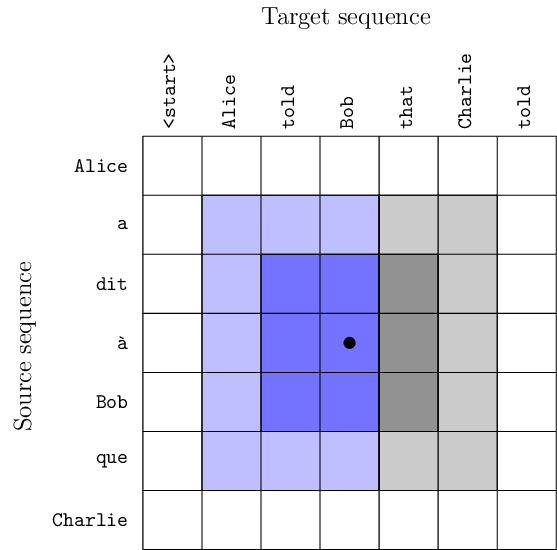
In [25] we investigate the limitations of the maximum likelihood estimation (MLE) used when training recurrent neural network language models. First, the MLE treats all sentences that do not match the ground truth as equally poor, ignoring the structure of the output space. Second, it suffers from "exposure bias": during training tokens are predicted given ground-truth sequences, while at test time prediction is conditioned on generated output sequences. To overcome these limitations we build upon the recent reward augmented maximum likelihood approach i.e., sequence-level smoothing that encourages the model to predict sentences close to the ground truth according to a given performance metric. We extend this approach to token-level loss smoothing, and propose improvements to the sequence-level smoothing approach. Our experiments on two different tasks, image captioning (see Fig. 23) and machine translation, show that token-level and sequence-level loss smoothing are complementary, and significantly improve results.

Pervasive Attention: 2D Convolutional Neural Networks for Sequence-to-Sequence Prediction
Participants : Maha Elbayad, Laurent Besacier [LIG] , Jakob Verbeek.
Current state-of-the-art machine translation systems are based on encoder-decoder architectures, that first encode the input sequence, and then generate an output sequence based on the input encoding. Both are interfaced with an attention mechanism that recombines a fixed encoding of the source tokens based on the decoder state. In [24], we propose an alternative approach which instead relies on a single 2D convolutional neural network across both sequences as illustrated in Figure 24. Each layer of our network re-codes source tokens on the basis of the output sequence produced so far. Attention-like properties are therefore pervasive throughout the network. Our model yields excellent results, outperforming state-of-the-art encoder-decoder systems, while being conceptually simpler and having fewer parameters.
|
Probabilistic Count Matrix Factorization for Single Cell Expression Data Analysis
Participant : Ghislain Durif.
The development of high-throughput biology technologies now allows the investigation of the genome-wide diversity of transcription in single cells. This diversity has shown two faces: the expression dynamics (gene to gene variability) can be quantified more accurately, thanks to the measurement of lowly-expressed genes. Second, the cell-to-cell variability is high, with a low proportion of cells expressing the same gene at the same time/level. Those emerging patterns appear to be very challenging from the statistical point of view, especially to represent and to provide a summarized view of single-cell expression data. PCA is one of the most powerful framework to provide a suitable representation of high dimensional datasets, by searching for latent directions catching the most variability in the data. Unfortunately, classical PCA is based on Euclidean distances and projections that work poorly in presence of over-dispersed counts that show drop-out events (zero-inflation) like single-cell expression data. In [22], we propose a probabilistic Count Matrix Factorization (pCMF) approach for single-cell expression data analysis, that relies on a sparse Gamma-Poisson factor model. This hierarchical model is inferred using a variational EM algorithm. We show how this probabilistic framework induces a geometry that is suitable for single-cell data visualization, and produces a compression of the data that is very powerful for clustering purposes. Our method is competed to other standard representation methods like t-SNE, and we illustrate its performance for the representation of zero-inflated over-dispersed count data. We also illustrate our work with results on a publicly available data set, being single-cell expression profile of neural stem cells. Our work is implemented in the pCMF R-package.
Extracting Universal Representations of Cognition across Brain-Imaging Studies
Participants : Arthur Mensch [Inria, Parietal] , Julien Mairal, Bertrand Thirion [Inria, Parietal] , Gael Varoquaux [Inria, Parietal] .
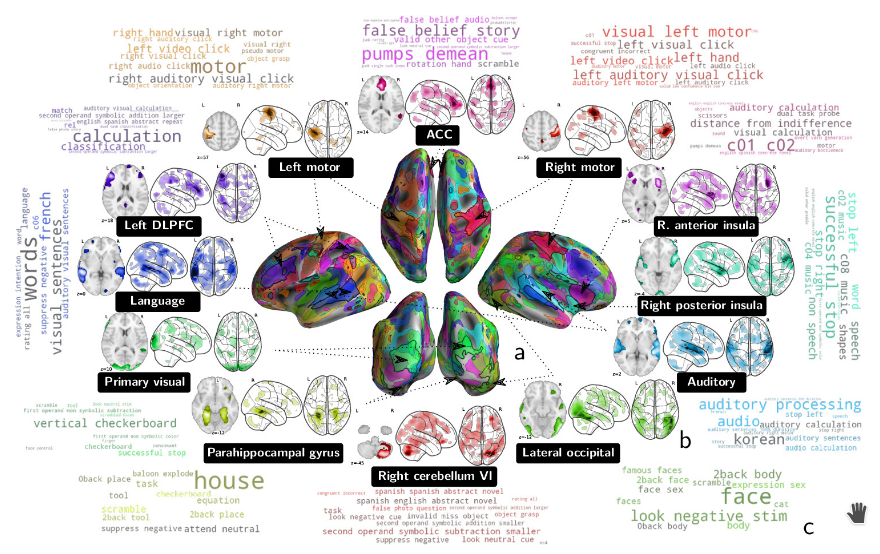
We show in [44] how to extract shared brain representations that predict mental processes across many cognitive neuroimaging studies. Focused cognitive-neuroimaging experiments study precise mental processes with carefully-designed cognitive paradigms; however the cost of imaging limits their statistical power. On the other hand, large-scale databasing efforts increase considerably the sample sizes, but cannot ask precise cognitive questions. To address this tension, we develop new methods that turn the heterogeneous cognitive information held in different task-fMRI studies into common-universal-cognitive models. Our approach does not assume any prior knowledge of the commonalities shared by the studies in the corpus; those are inferred during model training. The method uses deep-learning techniques to extract representations - task-optimized networks - that form a set of basis cognitive dimensions relevant to the psychological manipulations, as illustrated in Figure 25. In this sense, it forms a novel kind of functional atlas, optimized to capture mental state across many functional-imaging experiments. As it bridges information on the neural support of mental processes, this representation improves decoding performance for 80% of the 35 widely-different functional imaging studies that we consider. Our approach opens new ways of extracting information from brain maps, increasing statistical power even for focused cognitive neuroimaging studies, in particular for those with few subjects.
|
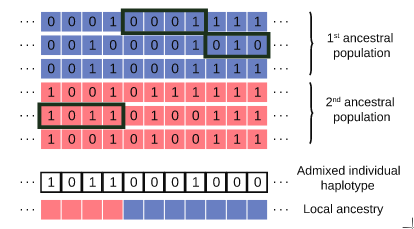
Loter: Inferring local ancestry for a wide range of species
Participants : Thomas Dias-Alves, Julien Mairal, Michael Blum [CNRS, TIMC Laboratory] .
Admixture between populations provides opportunity to study biological adaptation and phenotypic variation. Admixture studies can rely on local ancestry inference for admixed individuals, which consists of computing at each locus the number of copies that originate from ancestral source populations, as illustrated in Figure 26. Existing software packages for local ancestry inference are tuned to provide accurate results on human data and recent admixture events. In [5], we introduce Loter, an open-source software package that does not require any biological parameter besides haplotype data in order to make local ancestry inference available for a wide range of species. Using simulations, we compare the performance of Loter to HAPMIX, LAMP-LD, and RFMix. HAPMIX is the only software severely impacted by imperfect haplotype reconstruction. Loter is the less impacted software by increasing admixture time when considering simulated and admixed human genotypes. LAMP-LD and RFMIX are the most accurate method when admixture took place 20 generations ago or less; Loter accuracy is comparable or better than RFMix accuracy when admixture took place of 50 or more generations; and its accuracy is the largest when admixture is more ancient than 150 generations. For simulations of admixed Populus genotypes, Loter and LAMP-LD are robust to increasing admixture times by contrast to RFMix. When comparing length of reconstructed and true ancestry tracts, Loter and LAMP-LD provide results whose accuracy is again more robust than RFMix to increasing admixture times. We apply Loter to admixed Populus individuals and lengths of ancestry tracts indicate that admixture took place around 100 generations ago. The Loter software package and its source code are available at https://github.com/bcm-uga/Loter.
|