
2023Activity reportProject-TeamPLEIADE
RNSR: 201521167X- Research center Inria Centre at the University of Bordeaux
- In partnership with:CNRS, INRAE
- Team name: Patterns of diversity and networks of function
- In collaboration with:Laboratoire Bordelais de Recherche en Informatique (LaBRI), Biodiversité, Gènes & Communautés (BioGeCo)
- Domain:Digital Health, Biology and Earth
- Theme:Computational Biology
Keywords
Computer Science and Digital Science
- A3.1. Data
- A3.2. Knowledge
- A3.3.2. Data mining
- A3.3.3. Big data analysis
- A3.4. Machine learning and statistics
- A6.1. Methods in mathematical modeling
- A6.2. Scientific computing, Numerical Analysis & Optimization
- A8.2. Optimization
- A9.8. Reasoning
Other Research Topics and Application Domains
- B1.1.7. Bioinformatics
- B1.1.10. Systems and synthetic biology
- B3. Environment and planet
1 Team members, visitors, external collaborators
Research Scientists
- David Sherman [Team leader, INRIA, Senior Researcher, HDR]
- Clemence Frioux [INRIA, Researcher]
- Simon Labarthe [INRAE, Researcher]
Post-Doctoral Fellow
- Pablo Andres Ugalde Salas [INRIA, Post-Doctoral Fellow, until Oct 2023]
PhD Students
- Chabname Ghassemi Nedjad [UNIV BORDEAUX]
- Maxime Lecomte [INRIA, from Dec 2023]
- Maxime Lecomte [INRAE, until Nov 2023]
Technical Staff
- Leonard Brindel [INRIA, Engineer, from Oct 2023]
- Jean-Marc Frigerio [INRAE, Engineer, IR]
- Coralie Muller [INRIA, Engineer]
- Franck Salin [INRAE, Engineer, IEHC]
Interns and Apprentices
- Leonard Brindel [INRIA, Apprentice, until Jun 2023]
- Rafael Augusto Kaempfer Danin [INRIA, Intern, from Mar 2023 until Jun 2023]
Administrative Assistant
- Catherine Cattaert Megrat [INRIA]
Visiting Scientists
- Constanza Andreani Gerard [UNIV CHILI, from Oct 2023 until Nov 2023]
- Sebastien Raguideau [Earlham Institute]
External Collaborators
- Alain Franc [INRAE, from May 2023, HDR]
- Alain Franc [INRAE, until Apr 2023, HDR]
2 Overall objectives
The simplest ecosystem is a complex network of interactions between a diversity of organisms providing a diversity of functions. Each organism is the result of evolutionary processes driven by the creation then the selection of molecular diversity. Every function is a coordinated cascade of biochemical reactions, sensitive to substrate and environment, arising to adapt to the needs of the organism. Pleiade explores the diversity of organisms and the diversity of their functions and, as a fundamental challenge, seeks to formalize the links between them.
Pleiade measures the diversity of organisms by comparing DNA sequences and describes it using geometric methods. Amplicon sequences from metabarcoding are compared systematically to produce matrices of taxonomic distances. Mathematical analysis of these distance matrices, made possible by advances in dimension reduction, pattern recognition, and high-performance computing, reveals complex descriptions of the molecular diversity of the organisms in the sampled environment.
Pleiade examines the diversity of functions performed by these organisms by identifying the genes responsible for biochemical processes and grouping them into metabolic networks. Annotation of whole genome and metagenomic sequences allows us to delineate the functions provided by individual organisms and describe the interactions between them. Metabolic and process-based models are developed as compact descriptions of functional diversity. In addition to providing a means to simulate a system, a model is a syntactic object that symbolically represents a range of functional behaviors; patterns in the diversity of the encoded functions can be explored inferentially without exhaustively simulating the model to enumerate the set of behaviors it represents.
Comparison of annotated genomes and their associated metabolic networks reveals how functions arose over time: what functions, when they arose, and by which evolutionary mechanisms. Annotation is not performed on genomes individually, but comparatively, taking into account the similarities and differences between related species and strains.
A further challenge, developed recently, is considering the challenge of linking diversity and function in the particular context of microbial communities. We are developing a synergistic, iterative combination of a community-based strategy for deciphering the diversity in cultures and environmental samples, through metagenomic and metabolomic analysis of functional diversity and metabarcoding analysis of taxonomic diversity; and a function-based strategy for constructing digital twins of natural or designed communities through numerical models. The goal is a hybrid framework for studying systems dynamics using spatio-temporal models.
Shared methodologies needed to scale up to the complexity of biological systems, include high-performance computing (HPC); machine learning, including clustering, meta-modeling and classification for knowledge engineering; machine reasoning, specifically logical and rule-based methods used for model inference and network analysis. Logicial methods in particular promote explainable inference, since the rules are expressed in biological terms and are auditable by biologists, independently from the combinatorial and heuristic optimization techniques used to apply the rules.
Pleiade maintains strong collaborative relations with experimental biologists, and is committed to developing applications in ecology, evolution, biotechnology, and health. Team resources are dedicated to facilitating the adoption of our research by non specialist users, through development of reusable software, integration in HPC frameworks, improvement of web-based environments, and deployment of Jupyter, Galaxy, and Kubernetes interfaces.
3 Research program
Pleiade's mutually reinforcing strengths are a stable foundation of ecology and comparative genomics, and a novel synthesis of new methods extending our reach into microbial and planktonic communities and their dynamics. Our shared aim is to develop both: new challenges in microbial or planktonic communities leverage our solid expertise in foundational methods, and at the same time define new challenges for improving those foundations. We focus on reinforcing of a first set of disciplinary activities related to innovations in methods, be they in applied mathematics or in computer sciences, and a second set of interdisciplinary activities, to build advantageous assemblies of methods for understanding and managing biological systems of interest.
3.1 Research: A Geometric View of Diversity
Data analysis algorithms and tools must be revisited and scaled up. We mobilize both distributed algorithms and new algorithms, like random projection or column selection methods, to build point clouds in Euclidean spaces from massive data sets, and thus overcome the cubic complexity of computation of eigenvectors and eigenvalues of very large dense matrices. We also link distance geometry 51 with convex optimization procedures through matrix completion 34, 37.
Intercalibration: There is a considerable difference between supervised and unsupervised clustering: in supervised clustering, the result for an item is independent from the result for an item , whereas in unsupervised clustering, the result for an item (e.g. the cluster it belongs to, and its composition) depends on nearby items . Which means that the result may change if some items are added to or subtracted from the sample. This raises the more global problem of how to merge two studies to yield a more comprehensive view of biodiversity?
See 44 for some of our recent work linking the distance geometry problem, nonlinear mapping, and weighted least-squares scaling.
Project-team positioning
This research topic is about metabarcoding, i.e. producing inventories through classification, or producing OTUs with clustering, as elementary bricks for diversity, in a way analgous to the role that species play in morphological or molecular based taxonomy. The number of reads produced by NGS facilities is a challenge for bioinformatics and data analysis downstream into these directions: most algorithms scale with the square or the cube of the number of involved reads, which are counted in the millions. Most approaches look for heuristics permitting to produce result within a reasonable time (see e.g. UCLUST or USEARCH as wrappers for many solutions). The Pleiade team has explored another path: relying on HPC (the possibilities of which are underestimated in the community of diversity studies) to derive exact algorithms without heuristics, for computing distances between reads and to analyse distance matrices with dimension reduction.
This has been made possible due to an involvement in scientific computing, associating algorithms, their implemntation and optimisation. Such a bridge between scientific computing and characterisation of the biodiversity is original and still an exploratory field. It is develped in collaboration with project teams HiePACS, Storm and Tadaam in Inria Bordeaux, through two technology development actions: one, ADT Gordon, which is currently being valorized by the redaction of an article, and the second, Diodon, under development.
Collaborations
- In collaboration with the Pasteur Institute in Cayenne and the INRA MIA Research Team in Toulouse, Pleiade is developing a stochastic model for simulation of metacommunities, in the framework of patch occupancy models. The objective is a better understanding of zoonose propagation, namely rabies through bat hosts in connection with disturbances of pristine forests in French Guiana, which have an impact on the exposure of human populations to wildlife that act as reservoirs of zoonoses.
- We have co-supervised with Anne Lavergne (Institut Pasteur de Guyane) a PhD student at IPG and University of Cayenne (Sourakhata Tirera, defense on December 17, 2021) on the drivers of the diversity of viromes of rodents and birds. One part is about bioinformatics in order to select viral fragment, which are a minority in shotgun sequencing of viromes, and a second part is about disentangling the role of the habitat and of the phylogeny of the host in viral diversity. This has led to manuscript 60. A second one is in preparation to make available a new pipeline for dechimerisation of contings after shotgun sequencing and de nova assembly of reads into contigs (a process which is known to create many chimera).
- The Laboratory of excellence (Labex) CEBA promotes innovation in research on tropical biodiversity. It brings together a network of internationally-recognized French research teams, contributes to university education, and encourages scientific collaboration with South American countries. Pleiade has participated in three current international projects funded by CEBA:
- MicroBIOMES: Microbial Biodiversities
- Neutrophyl: Inferring the drivers of Neotropical diversification
- Phyloguianas: Biogeography and pace of diversification in the Guiana Shield
- We collaborate with Institut Pasteur de Guyane at Cayenne for developing the domain of so-called Ecoviromics for some zoonoses in French Guiana. On top of co-supervizing a PhD student at IPG in Cayenne as mentioned above (deciphering the respective roles of host phylogeny and environmental variables in the virome of different hosts (bats, rodents, birds), this collaboration has led to a participation in a synthesis paper on Ecology, evolution, and epidemiology of zoonotic and vector-borne infectious diseases in French Guiana 59.
3.2 Research: Community-scale Metabolic and Omics-based Modeling
Metabolism can be abstracted into sets of metabolic reactions, associated to the genome through gene-protein relationships, and connecting substrates to products thereby forming metabolic networks. Genome-scale metabolic networks (GSMNs) contain all the reactions predicted to occur in a organism according to its genomic contents. Combined with additional knowledge on the system, possibly other –omics data and mathematical models, GSMNs are used to predict the behaviour of an organism or a community of organisms.
A widely-used mathematical formalism for modeling GSMNs is constraint-based modeling, among which flux balance analysis (FBA) is the main representative 53. Such methods permit a quantitative prediction of activity fluxes in metabolic networks while optimising an objective function and assuming steady-state of the system.
The emergent metabolism of microbial communities can also be qualitatively modeled using a boolean approximation of metabolic dynamics8. In this approach the behavior of the system is described by logical rules that activate a given reaction as soon as its substrates become available; numerical parameters such as stochiometry or enzyme kinetics are ignored in favor of graph topology and paths. The advantage is that such qualitative models, unlike quantitative methods such as FBA, do not require the assumption that the system is at steady-state and can model systems where cells are constantly growing or constantly reproducing.
Network expansion, introduced in 43 as a recursive traversal of the structure of a metabolic graph, lends itself to concise definition using answer set programming (ASP) 49 and thus to efficient implementation using SAT solvers 48. In practice, using ASP for metabolic modeling makes it possible to define both the activation of metabolic reactions in different conditions, and the constraints and optimizations needed to find solutions in a combinatorically large state space.
We focus in particular on the key question of determining minimal communities, subsets of the organisms present in an environment that are sufficient to reproduce a chosen behavior 45. The methodological goal here is to identify key species in a community through use of ASP to rapidly explore the search space and thus, through heuristic resolution of combinatorial problems, provide the guarantees an exhaustive search with a greatly reduced computational cost 4.
Functional and taxonomic diversities, beyond intrinsic specificities encoded in the genetic material, are also strongly shaped by their environment. Spatial nutritional niches, microbial interactions and abiotic constraints lead to complex spatial structures in the microbial community that impact its overall dynamics. PDE-based models of the microbiota in its environment allow including in the model these multiple mechanisms in order to decipher their influence on the community faith.
The main methodological developments in this area are related to mathematical modeling (in particular the correct level of simplification in the multi-physics description of the microbial environment), model simplification (asymptotic approximation), inference from multi-omics data (including dimension reduction, statistical learning) and numerical developments (in particular fast approximation of metabolic models with machine learning methods). Strong interactions with community-scale metabolic models are sought, specially for multi-omics inference and knowledge-based machine learning constraints.
The goal is to achieve accurate models of microbial communities that could be used as digital twins of controlled experiments in microbial ecology. Culturomic facilities allow for the acquisition of multi-omics time-series data in controlled conditions, which can be used to build and fit population dynamics models, and can be used in turn to explore numerically biological assumptions and to help in experimental planning and data analysis.
Project-team positioning
The team has expertise in the state of the art methods for metabolic modeling and in metabolic network reconstruction through earlier works 45, 46, 32, 54. The team also masters ODE or PDE microbial population dynamics models including their complex environment 36, 50 and parameter inference with experimental data 55. We work with international or national teams (see below) for combinatorial problem solving (University of Potsdam, Germany), computational biology for health (Quadram Institute Bioscience, UK) and biological applications (Roscoff Biological Station, INRAE teams).
Other international teams working on the subject of community metabolic modeling include but is not restricted to the research groups of: Ines Thiele (U. Galway, Ireland), Kiran Patil (U. Cambridge, UK), Karoline Faust (U. Leuven, Belgium), Daniel Machado (Norwegian University of Science and Technology).
Among Inria project-teams, Dyliss (Rennes) is the one with the closest research themes. Clémence Frioux did her PhD in this team (2015-2018) and stayed for an additional 6 months after the defense. As a result, the majority of her past contributions were done in collaboration with Dyliss members, and current collaborations persist through co-development and maintenance of software, applications etc.
Collaborations
- Quadram Institute Bioscience and Earlham Institute: meta-analyses of metagenomic cohorts for the human gut microbiota.
- University of Potsdam: answer set programming and combinatorial problem solving.
- Station Biologique de Roscoff: algae applications.
- INRAE BFP, STLO, MaIAGE, SAVE, Micalis, IEES: applications and methodological development in computational biology and mathematics.
- CEA. Bio-inspired digital sensors in the framework of the Pherosensor project.
- U. Besançon and U. Orléans. Modeling of the fluidic environment in the gut microbiota.
- U. Paris-Saclay/U.Evry. Machine learning with ANOVA-RKHS.
- Inria Bretagne Atlantique: systems biology and systems ecology.
- Ysopia Bioscience by means of an Inria Tech contract focused the analysis of metagenomic data and the connection to the metabolic screening provided by Metage2Metabo.
- Biomathematica through a CIFRE PhD hosted at MaIAGE (INRAe) co-supervised by Pleiade.
3.3 Research: Bioinformatics, Genomes, and Knowledge Management
The heterogenous data generated in computational molecular biology and ecology are distinguished not only by their volume, but by the richness of the many levels of interpretation that biologists create. The same nucleic acid sequence can be seen as a molecule with a structure, a sequence of base pairs, a collection of genes, an allele, or a molecular fingerprint. To extract the maximum benefit from this treasure trove we must organize the knowledge in ways that facilitate extraction, analysis, and inference. Our focus has been on the efficient representation of relations between biological objects and operations on those representations, in particular heuristic analyses and logical inference.
Pleiade developes applications in comparative genomics of related organisms, using novel mathematical tools for representing compactly, at different scales of difference, comparisons between related genomes. New methods based on distance geometry will refine these comparisons. Compact representations can be stored, exchanged, and combined. They form the basis of novel simultaneous genome annotation methods, that can be linked directly to abductive inference methods for building functional models of the organisms and their communities.
Since a goal of Pleiade is to integrate diversity throughout the analysis process, it is necessary to incorporate diversity as a form of knowledge that can be stored in a knowledge base. Diversity can be represented using various compact representations, such as trees and quotient graphs storing nested sets of relations. Extracting structured representations and logical relations from integrated knowledge bases requires domain-specific query methods that can express forms of diversity.
Project-team positioning
Historically, Pleiade members have been pioneers in the development of large-scale eukaryote comparative genomics. We were involved since the late 1990's in the first genome sequencing of eukaryote microorganisms, co-authored 8 articles in the 30 special issue presenting the first large-scale comparative genomics study, and were in the first authors of the landmark Nature article 42 comparing five complete annotated genomes. Our articles in comparative genomics, particularly of the hemiascomycetous yeasts of biotechnological interest, have achieved thousands of citations and continue to do so decades later. A principle that we fought for 39, 58, that comparative genomics must be based on a systematic and mathematical comparison of the genomes, rather than on an opportunistic one-against-all comparison to the model organism du jour, is now considered standard practice. We also originally set the standard for web-based tools for comparative genomics, organized around the principle of an interaction design based on the questions asked by by biological user, rather than based on the organization of the underlying database as seen by a computer scientist 57, 58. Pleiade capitalized on this experience in support of the research efforts describe above, and also through a wide network of collaborators in the biological sciences. Our current work applies the principles of comparative genomics to a series of smaller projects focused on collections of genomes of biotechnological or of health-related interest.
Collaborations
- Institut des Sciences du Vigne et du Vin (ISVV), U. Bordeaux
- Vitapalm (LEAP-Agri)
- Laboratoire de Microbiologie Fondamentale et Pathogénicité (LMFP), UMR 5234 CNRS U. Bordeaux
- Laboratory of Membrane Biology (LBM) UMR 5200 CNRS U. Bordeaux
4 Application domains
4.1 Molecular based systematics and taxonomy
Defining and recognizing the myriads of species occuring in the biosphere has been the focus of phenomenal energy over the past centuries and remains a major goal of Natural History. It is an iconic paradigm in pattern recognition (clustering has coevolved with numerical taxonomy many decades ago). Developments in evolution and molecular biology, as well as in data analysis, have over the past decades enabled a profound revolution, where species can be delimited and recognized by data analysis of sequences. We aim at proposing new tools, in the framework of E-science, which make possible () better exploration of the diversity in a given clade, and () assignment of a place in these patterns for new, unknown organisms, using information provided by sets of sequences. This will require investment in data analysis, machine learning, and pattern recognition to deal with the volumes of data and their complexity.
One example of this project is about the diversity of trees in Amazonian forest, in collaboration with botanists in French Guiana. Protists (unicellular Eukaryots) are by far more diverse than plants, and far less known. Molecular exploration of Eukaryotes diversity is nowadays a standard in biodiversity studies. Data are available, through metagenomics, as an avalanche and make molecular diversity enter the domain of Big Data. Hence, an effort will be invested, in collaboration with other INRIA teams (GenScale, HiePACS) for porting to HPC algorithms of pattern recognition and machine learning, or distance geometry, for these tools to be available as well in metagenomics. This will be developed first on diatoms (unicellular algae) in collaboration with INRAE team at Thonon and University of Uppsala, on pathogens of tomato and grapewine, within an existing network, and on bacterial communities, in collaboration with University of Pau. For the latter, the studies will extend to correlations between molecular diversity and sets of traits and functions in the ecosystem.
4.2 Genome and transcriptome annotation
Sequencing genomes and transcriptomes provides a picture of how a biological system can function, or does function under a given physiological condition. Simultaneous sequencing of a group of related organisms is now a routine procedure in biological laboratories for studying a behavior of interest, and provides a marvelous opportunity for building a comprehensive knowledge base of the relations between genomes1, 12. Key elements in mining these relations are: classifying the genes in related organisms and the reactions in their metabolic networks, recognizing the patterns that describe shared features, and highlighting specific differences.
Pleiade develops applications in comparative genomics of related organisms, using new mathematical tools for representing compactly, at different scales of difference, comparisons between related genomes. New methods based on computational geometry refine these comparisons. Compact representations can be stored, exchanged, and combined. They will form the basis of new simultaneous genome annotation methods, linked directly to abductive inference methods for building functional models of the organisms and their communities.
Our ambition in biotechnology is to permit the design of synthetic or genetically selected organisms at an abstract level, and guide the modification or assembly of a new genome. Our effort is focused on two main applications: genetic engineering and synthetic biology of oil-producing organisms (biofuels in CAER, palm oils), and improving and selecting starter microorganisms used in winemaking (collaboration with the ISVV and the BioLaffort company).
4.3 Community ecology
Community assembly models how species can assemble or diassemble to build stable or metastable communities. It has grown out of inventories of countable organisms. Using metagenomics one can produce molecular based inventories at rates never reached before. Most communities can be understood as pathways of carbon exchange, mostly in the form of sugar, between species. Even a plant cannot exist without carbon exchange with its rhizosphere. Two main routes for carbon exchange have been recognized: predation and parasitism. In predation, interactions–even if sometimes dramatic–may be loose and infrequent, whereas parasitism requires what Claude Combes has called intimate and sustainable interactions 38. About one decade ago, some works 56 have proposed a comprehensive framework to link the studies of biodiversity with community assembly. This is still incipient research, connecting community ecology and biogeography.
We aim at developping graph-based models of co-occurence between species from NGS inventories in metagenomics, i.e. recognition of patterns in community assembly, and as a further layer to study links, if any, between diversity at different scales and community assemblies, starting from current, but oversimplified theories, where species assemble from a regional pool either randomly, as in neutral models, or by environmental filtering, as in niche modeling. We propose to study community assembly as a multiscale process between nested pools, both in tree communities in Amazonia, and diatom communities in freshwaters. This will be a step towards community genomics, which adds an ecological flavour to metagenomics.
Next-generation sequencing technologies are now an essential tool in population and community genomics, either for making evolutionary inferences or for developing SNPs for population genotyping analyses. Two problems are highlighted in the literature related to the use of those technologies for population genomics: variable sequence coverage and higher sequencing error in comparison to the Sanger sequencing technology. Methods are developed to develop unbiased estimates of key parameters, especially integrating sequencing errors 52. An additional problem can be created when sequences are mapped on a reference sequence, either the sequenced species or an heterologous one, since paralogous genes are then considered to be the same physical position, creating a false signal of diversity 47. Several approaches were proposed to correct for paralogy, either by working directly on the sequences issued from mapped reads 47 or by filtering detected SNPs. Finally, an increasingly popular method (RADseq) is used to develop SNP markers, but it was shown that using RADseq data to estimate diversity directly biases estimates 33. Workflows to implement statistical methods that correct for diversity biases estimates now need an implementation for biologists.
5 Social and environmental responsibility
5.1 Footprint of research activities
Pleiade's policy is to rely on shared computing platforms for computations that consume significant energy, for two reasons. First, those platforms have greater leverage over total energy consumption, and have the technical means to implement green computing on a useful scale. Second, those platforms have staff with the requisite skills to develop and implement policies as part of their service offer. Our partner platforms are mesocenters in Bordeaux and Grenoble, and national centers including the Idris and the CEA. Some of them charge back the cost of CO generation. The scientific committee of the Bordeaux mesocenter MCIA, which has a member from Pleiade, actively debates green computing implementation. Pursuant to our team policy, Pleiade does not use high-powered workstations on the desktop.
5.2 Promoting equality and diversity in science
Promoting the inclusion and diversity in science is essential at all levels: when planning science during project conception, when executing science and publishing its results, and also within the community of scientists itself. Members of Pleiade are involved in promoting the place of women in science through the participation in outreach activities, but also by committing to working groups and committees on the subject at the local and national level.
6 Highlights of the year
- A landmark large-scale meta-analysis of 5,278 adult and infant fecal metagenomes at three levels — human, household, and geographical region — revealed that heredipersistent microbes owe much of their persistence to constant reinfections from the environment, and their presence may in turn be important for the next generation. In consequence, current medical procedures for the human gut microbiome, such as microbiota transplants, may be futile or even harmful. 15
- Integrating anaerobic microbiology knowledge with statistical learning can focus metagenomic analysis on functional profiles in microbiota. Applying this approach to fibre degradation in the gut identified four distinct functional profiles that can be easily monitored as markers of diet, dysbiosis inflammation and disease. 16
- MISTIC (§10.3.1), a flagship project coordinated by Pleiade of the Programme et équipements prioritaires de recherche (PEPR) Agroecology and ICT, kicked off in March 2023. MISTIC will elucidate the role of microbial community dynamics in the adaptation of crop plants to environmental stresses including climate change.
7 New software, platforms, open data
7.1 New software
7.1.1 Metage2Metabo
-
Keywords:
Metabolic networks, Microbiota, Metagenomics, Workflow
-
Scientific Description:
Flexible pipeline for the metabolic screening of large scale microbial communities described by reference genomes or metagenome-assembled genomes. The pipeline comprises several main steps. (1) Automatic and parallel reconstruction of metabolic networks. (2) Computation of individual metabolic potentials (3) Computation of collective metabolic potential (4) Calculation of the cooperation potential described as the set of metabolites producible by species only in a cooperative context (5) Computation of minimal-sized communities sastifying a metabolic objective (6) Extraction of key species (essential and alternative symbionts) associated to a metabolic function
-
Functional Description:
Metabolic networks are graphs which nodes are compounds and edges are biochemical reactions. To study the metabolic capabilities of microbiota, Metage2Metabo uses multiprocessing to reconstruct metabolic networks at large-scale. The individual and collective metabolic capabilities (number of compounds producible) are computed and compared. From these comparisons, a set of compounds only producible by the community is created. These newly producible compounds are used to find minimal communities that can produce them. From these communities, the keytstone species in the production of these compounds are identified.
- URL:
- Publication:
-
Contact:
Clemence Frioux
-
Participants:
Clemence Frioux, Arnaud Belcour, Anne Siegel
7.1.2 MiSCoTo
-
Name:
Microbiota Screening and COmmunity Selection with TOpology
-
Keywords:
Metabolic networks, ASP - Answer Set Programming, Logic programming
-
Scientific Description:
MiSCoTo solves combinatorial problems using Answer Set Programming. It aims at minimizing either the number of selected species or both the number of selected species and the cost of the interaction between them, characterized by the number of metabolic exchanges. In the first case, the level of modeling is called lumped or mixed-bag, in the latter, it is compartmentalized.
-
Functional Description:
Metabolic networks are composed of biochemical reactions and gather the expected metabolic capabilities of species. For organisms that live in interaction altogether (microbiotas), complementarity between these networks can be exploited to predict cooperation events. This software takes as inputs metabolic networks for various species (host, symbionts of the microbiota), components of the growth medium and a metabolic objective (metabolites to be produced), and aims at selecting a minimal set of symbionts to ensure the metabolic objective can be achieved. The software can use two types of modelings: a simplified one and another that takes into account the cost of metabolic exchanges and aims at minimizing it.
-
Release Contributions:
Memory usage optimization. Fix issues with input file formats.
- URL:
- Publication:
-
Contact:
Clemence Frioux
-
Participants:
Clemence Frioux, Anne Siegel, Enora Fremy, Camille Trottier, Arnaud Belcour
7.1.3 MeneTools
-
Name:
Metabolic networks Topological tools
-
Keywords:
Metabolic networks, Graph, Topology, Bioinformatics, Systems Biology, ASP - Answer Set Programming
-
Scientific Description:
MeneTools are a set of tools for the exploration of the producibility potential in a metabolic network using the network expansion algorithm. The MeneTools can: - assess whether targets are producible starting from nutrients (Menecheck) - get all compounds that are producible starting from nutrients (Menescope) - get all reactions that are activable from nutrients (Meneacti) - get production paths of specific compounds (Menepath) - obtain compounds that if added to the nutrients, would ensure the producibility of targets (Menecof) - identify metabolic deadends, i.e. metabolites that act as reactants of reactions but never as products, or metabolites that act as products of reactions but never as reactants. This is a purely structural analysis. All MeneTools using modelling follow the producibility in metabolic networks as defined by the network expansion algorithm.
-
Functional Description:
MeneTools consist in four topological tool to analyze metabolic models in a graph-based perspective. Menecheck verifies the producibility of target compounds from available substrates (growth medium) of the metabolic network. Menescope gives the whole range of accessible compounds in the metabolic network starting from substrates. Menepath give the production paths of given compounds in the model. Menecof proposes compounds that need to be produced or added as substrate for ensuring the producibility of targets.
- URL:
- Publications:
-
Contact:
Clemence Frioux
-
Participants:
Clemence Frioux, Anne Siegel, Arnaud Belcour
7.1.4 Emapper2GBK
-
Keywords:
Bioinformatics, Metabolic networks, Functional annotation
-
Functional Description:
Starting from FASTA and Eggnog-mapper annotation files, Emapper2GBK builds a GBK file that is suitable for metabolic network reconstruction with Pathway Tools, and adds the GO terms and EC numbers annotations in the GenBank file.
- URL:
- Publication:
-
Contact:
Clemence Frioux
-
Participants:
Clemence Frioux, Arnaud Belcour, Anne Siegel
7.1.5 Biodiversiton
-
Name:
Biodiversiton
-
Keywords:
Biodiversity, Comparative metagenomics, Clustering, Dimensionality reduction, Masses of data
-
Functional Description:
Biodiversiton is a suite of tools for biodiversity composed by Rsyst, pairwise_dis, diagno_syst, and yapotu. The global project provides tutorials, datasets, and a readme for the whole suite.
- URL:
-
Authors:
Alain Franc, Jean-Marc Frigerio, Franck Salin
-
Contact:
Alain Franc
7.1.6 Yapotu
-
Name:
Yet Another Pipeline for OTU building
-
Keywords:
Metagenomics, Biodiversity, Dimensionality reduction, Masses of data
-
Functional Description:
The main functionalities are as follows: 1) building OTUs from a fasta file (swarm, vsearch, ..) or a distannce file (yapotu) for an environmenal sample 2) building a fasta file and a distance file per OTU 3) checking the consistency of the OTUs by displaying them as a graph (see OTU as a graph below) 4) displaying the shape of an OTU or of a set of OTUs by Multidimensional Scaling 5) implementing Hierachical Aggregative Clustering of an OTU or a set of OTUs with various aggregation methods
- URL:
-
Authors:
Alain Franc, Jean-Marc Frigerio, Franck Salin
-
Contact:
Alain Franc
-
Partner:
INRAE
7.1.7 pydiodon
-
Name:
pydiodon
-
Keywords:
Dimensionality reduction, Data analysis
-
Functional Description:
Most of dimension reduction methods inherited from Multivariate Data Analysis, and currently implemented as element in statistical learning for handling very large datasets (the dimension of spaces is the number of features) rely on a chain of pretreatments, a core with a SVD for low rank approximation of a given matrix, and a post-treatment for interpreting results. The costly part in computations is the SVD, which is in cubic complexity. Diodon is a list of functions and drivers which implement (i) pre-treatments, SVD and post-treatments on a large diversity of methods, (ii) random projection methods for running the SVD which permits to bypass the time limit in computing the SVD, and (iii) an implementation in C++ of the SVD with random projection at prescribed rank or precision, connected to MDS.
-
Release Contributions:
- completed documentation with sphynx - library now public through Inria git - availability of a readme - making a few "toy" datasets available - delivering a few jupyter notebooks as tutorials
- URL:
-
Authors:
Alain Franc, Florent Pruvost, Romain Peressoni, Romain Peressoni
-
Contact:
Alain Franc
7.1.8 TANGO
-
Keywords:
Computational biology, Systems Biology, Metabolic networks, Bacterial strains
-
Functional Description:
The organoleptic properties that provide the added value of fermented dairy products result from specific metabolites that are produced by metabolic processes performed in concert by consortia of microbial species. TANGO enable a deeper understanding of the molecular and cooperative mechanisms underlying the production of organoleptic compounds. Tango uses a combination of whole-genome metabolic modeling and dynamic numerical simulation to assemble a complete, precise model of cheese production using lactic acid and propionic acid bacteria. The results of this modeling reveal interactions between the members of the bacterial community, follow dynamically organoleptic compounds and fit with experimental data.
-
Contact:
Maxime Lecomte
7.1.9 Mapler
-
Name:
Metagenome Assembly and Evaluation Pipeline for Long Reads
-
Keywords:
Metagenomics, Genome assembly, Benchmarking, Bioinformatics
-
Functional Description:
Mapler is a pipeline to compare the performances of long-read metagenomic assemblers. The pipeline is focused on assemblers for high fidelity long read sequencing data (e.g. pacBio HiFi), but it supports also assemblers for low-fidelity long reads (ONT, PacBio CLR) and hybrid assemblers. It currently compares metaMDBG, metaflye, Hifiasm-meta, opera-ms and miniasm as assembly tools, and uses reference-based, reference-free and binning-based evalutation metrics. It is implemented in Snakemake.
- URL:
- Publication:
-
Contact:
Nicolas Maurice
-
Participants:
Nicolas Maurice, Claire Lemaitre, Riccardo Vicedomini, Clemence Frioux
7.1.10 seed2lp
-
Keywords:
ASP - Answer Set Programming, Metabolic networks, Logic programming, Linear programming
-
Functional Description:
Seed2lp is a tool for seed searching from metabolic networks using logic and/or linear programming (reasoning, FBA or hybrid). The solution, developed in python, uses Answer Set programming with clingo and clingo-lpx, and allows solution verification with Cobrapy.
-
Contact:
Clemence Frioux
7.1.11 GeMeNet
-
Name:
Genomes to Metabolic Networks
-
Keywords:
Bioinformatics, HPC, Metabolic networks, Genomics
-
Scientific Description:
GeMeNet is a pipeline for generating multiple metabolic networks essentially from their genomes but from other data (gbk, ect...).
-
Functional Description:
GeMeNet is a pipeline for generating multiple metabolic networks essentially from their genomes but from other data (gbk, ect...).
- URL:
-
Authors:
Coralie Muller, Clemence Frioux
-
Contact:
Coralie Muller
7.1.12 CoCoMiCo
-
Name:
Cooperation and competition potentials in large microbial communities
-
Keywords:
Automated Reasoning, Metabolic networks, Answer Set Programming, Microbiota, Systems Biology
-
Scientific Description:
By discretely modelling metabolic cross-feeding and dependency on limiting metabolites between organisms, CoCoMiCo defines novel optimisation criteria that can be used at scale for screening microbial communities using combinatorial methods. The criteria can be used for evaluation of large sets of naturally-occurring communities, or of large sets of generated candidate communities screened to identify species of interest for health or ecology applications.
-
Functional Description:
Metabolic cross-feeding and dependency on limiting metabolites between organisms are logically modeled using an ad hoc knowledge base derived from whole genome metabolic models in SBML format, and analyzed by logical inference rules defined using the answer set programming paradigm.
- URL:
-
Contact:
David James Sherman
7.1.13 MetagenoPIC
-
Name:
Metagenomic pipeline creation
-
Keywords:
Metagenomics, Genome assembly
-
Scientific Description:
MetagenoPIC pipeline contain differents steps in order to reconstruct Metagenome Assembled Genomes (MAGs).
The first step of pipeline is read trimming using kneaddata, with read filtering against organism databases. The next step is assembly step, using either Megahit or metaSpades. Reads are aligned to these contigs using BWA-MEM2. The following step is binning: the pipeline groups contigs into bins using one of two ways. The first way is to use a single one of the MetaBAT2, MaxBin2, or CONCOCT tools. The second way is to run a combination of these tools and then concatenate the results using a bin refiner, either DASTool or MetaWrap. The next step is to dereplicate the resulting bins, filtering reads of poor quality (high contamination and low completeness) and dereplicating bins that can be represent the same metagenome. In all cases, a threshold for the contamination and completeness can be specified.
Once the pipeline has constructed MAGs, it can run further analyses: CheckM to evaluate the quality of bins, and GTDB-TK to run a taxonomic assignment.
-
Functional Description:
Metagenome reconstruction comprises four steps: assembly, binning, dereplication, annotation.
-
Release Contributions:
Internal availability
-
News of the Year:
The first internal release of MetagenoPIC based on work by Ariane Badoual provides a full pipeline that can be executed by any system that respects the Common Workflow Language. Validation was performed using Calrissian on Kubernetes and Toil on slurm. Work by Leonard Brindel this year included integration of long reads, improvements to functional and structural annotation using Prodigal and Eggnog-mapper, and an overall increase in reliability.
- URL:
-
Contact:
Clemence Frioux
-
Participants:
Ariane Badoual, Clemence Frioux, David Sherman, Leonard Brindel
7.2 New platforms
Participants: David Sherman, Ahmed Kallel.
As a founding principle, Pleiade supports reproducible scientific analyses and promotes a declarative approach using reusable software modules, rigorous documentation of data provenance, and systematic recording of workflows. The latter is a challenge when interactive interfaces are used, but can be addressed, to cite two examples, in Galaxy by extracting workflows, and in other systems by using Jupyter notebooks. Part of Pleiade's mission is to automate the deployment of environments that support these goals, for non-technical end users.
Pleiade has built a Kubernetes platform Pleiadès hosted in the Inria research center at the Univ. Bordeaux, which in 2023 was been integrated into Inria's IT Management (DSI-SP).
Use cases were identified by the project-team and from the MISTIC data management plan:
- Fast deployment of containerized user environments, combining biological data and databases, software modules specified by version, a CWL executor, and interactive tools including web front ends, notebooks, or Galaxy. A user environment will provide at least one specific HTTPS endpoint, created dynamically. A single researcher may deploy several different environments in the course of one day.
- Support for development and testing of workflows, as above but configured for team members who are developing software modules or interfaces, and who must often deploy several different environments simultaneously.
- Dynamically allocated containerized compute tasks, including both individual analysis steps in workflows and GitLab runner containers used for continuous integration. These tasks arrive in bursts that often cannot be planned in advance.
- Long-running stream preprocessing, a low-priority background task that watches external databases for changes, chooses pertinent data, precomputes representations and ingests them into local data bases.
The following requirements were derived from these use cases:
- Tasks must run in OCI containers. A typical environment will be constructed from ten to one hundred containers, grouped in Kubernetes Pods of co-localized containers that share a private network.
- Containers run unprivileged and must rely on role-based access control (RBAC), secrets, and service accounts.
- Different storage classes must be available for dynamic volume allocation: ReadWriteSingle, ReadWriteMany, Object (S3) Bucket.
- An application must be able to allocate a route with wildcard DNS in order to offer an endpoint, internally to the Inria network.
- A collection of Kubernetes custom resource definitions and RBAC definitions, specific to Pleiade's applications, is needed.
- A collection of OpenShift Operators for deployment of applications, is needed. These include database services, workflow execution, and container building using source-to-image (S2I).
- A management interface through the OKD console that allows inspection and management of app topologies, pods, volumes, and Kubernetes objects.
We support community best practices for reproducible computing in bioinformatics, using biocontainers generated by bioconda, in CWL or Galaxy workflows. For internal use we provide TES endpoints and host JupyterHub environments.
The Pleiadès platform is built on OKD 4, the community distribution of Kubernetes developed alongside of RedHat Openshift. OKD4 in particular uses the CRI-O runtime, not Docker, and containers run unprivileged. Software-defined storage and S3 endpoints are provided by Ceph. Pleiadès follows the gitops pattern and all management and implementation use Git repositories as the single source of truth.
7.3 Open data
Pleaide is strongly committed to open data and to providing machine-readable and reusable provenance with research outputs.
In the MISTIC data management plan, Pleiade and its partners specifically agreed to the following:
- Adoption of FAIR and Plan S principles.
- Data acquired by the project and provided as a deliverable will be deposited in reliable third-party archives such as data.gouv.fr.
- Software developed by the project will be publically hosted on gitlab.inria.fr during development.
- Software provided as a deliverable will be deposited in a reliable third-party archive such as inria.hal.science and Software Heritage, and made available under an open source license.
- Computational results provided in support of a published method or as a project deliverable will be made available in the form of a RO-Crate, providing complete, reproducible provenance.
Mutualised technical support for executing workflows with provenance is provided to Pleiade and its projects on the Pleiadès Kubernetes platform (§7.2).
8 New results
8.1 Modeling metabolism of microbial communities in time and space
Participants: Pablo Ugalde-Salas, Simon Labarthe, Clémence Frioux, Coralie Muller, Rafael Augusto Kaempfer Danin, Maxime Lecomte, David Sherman.
Metabolic models allow the prediction of the metabolic behaviour of microbial strains from the knowledge of their genomes. Quantitative methods such as Flux Balance Analysis provide a framework to compute metabolic fluxes (i.e. consumption and production rates of metabolites present in the environment of the microbe) and biomass growth at the cost of an optimization problem. When modeling the dynamics of microbial communities, a FBA model must be solved for each microbial strain of the community and at each time step of the dynamics, representing an important computational load.
In 2021, an Inria Exploratory Action - SLIMMEST – carried out by Simon Labarthe and Clémence Frioux – was initiated. This project aims at combining discrete reasoning models of metabolism to numerical metamodels and PDEs for the simulation of microbial communities in time and space. The selected methodology is the coupling between PDE-based microbial population dynamics model with metamodels of complex optimizations predicting their metabolism. The main difficulty here is to ensure the scalability of the simulation and the selection of relevant metabolic functions and species to be tracked over time. The grant permitting hiring a postdoc researcher (Pablo Ugalde) and an engineer (Coralie Muller). The aim of the project is to articulate qualitative methods to analyse the functional potential of a microbial community by analysing and simplifying the community-wide metabolic capabilities, to surrogate models of the individual FBA models to provide fast and accurate simulations of the community dynamics. Qualitative methods include Answer Set Programming (ASP) to predict metabolic interactions in the community, metabolic exploration and simplifications. Surrogate models are obtained with a statistical method named ANOVA-RKHS that build a specific RKHS allowing for feature selection and promissing trade-off between speed and accuracy.
In 2023, the work achieved during the 2021 CEMRACS project was published in the CEMRACS proceedings 18. Figure 1 illustrates the use of surrogate metamodels learned by ANOVA-RKHS to speed up dynamic flux balance analysis. It was applied to a microbial community consisting of a pathogen and a commensal of the human gut microbiome. We pursued the efforts in enhancing the models by including an metabolic model of human epithelial cells interacting with the microbiota-derived chemicals diffusing in the crypt lumen. The team hosted Rafael Augusto Kaempfer Danin as intern to work on the surrogate of the epithelial cell. By increasing the speed of dynamic simulations, the latest results make possible the construction of a spatialised model of the system that would otherwise not be tractable. Figure 2 describes the biological system that is modelled and the global methodology that is followed.
The project results have been presented by Coralie Muller and Pablo Ugalde at an international conferences and an international workshop: 31st Annual Intelligent Systems For Molecular Biology and the 22nd Annual European Conference on Computational Biology (ISMB/ECCB 2023, Lyon, France – poster) 26, 7th International Symposium on Systems Biology of Microbial Infection (SBMI 2023, Jena, Germany – talk and poster) 29, 27.
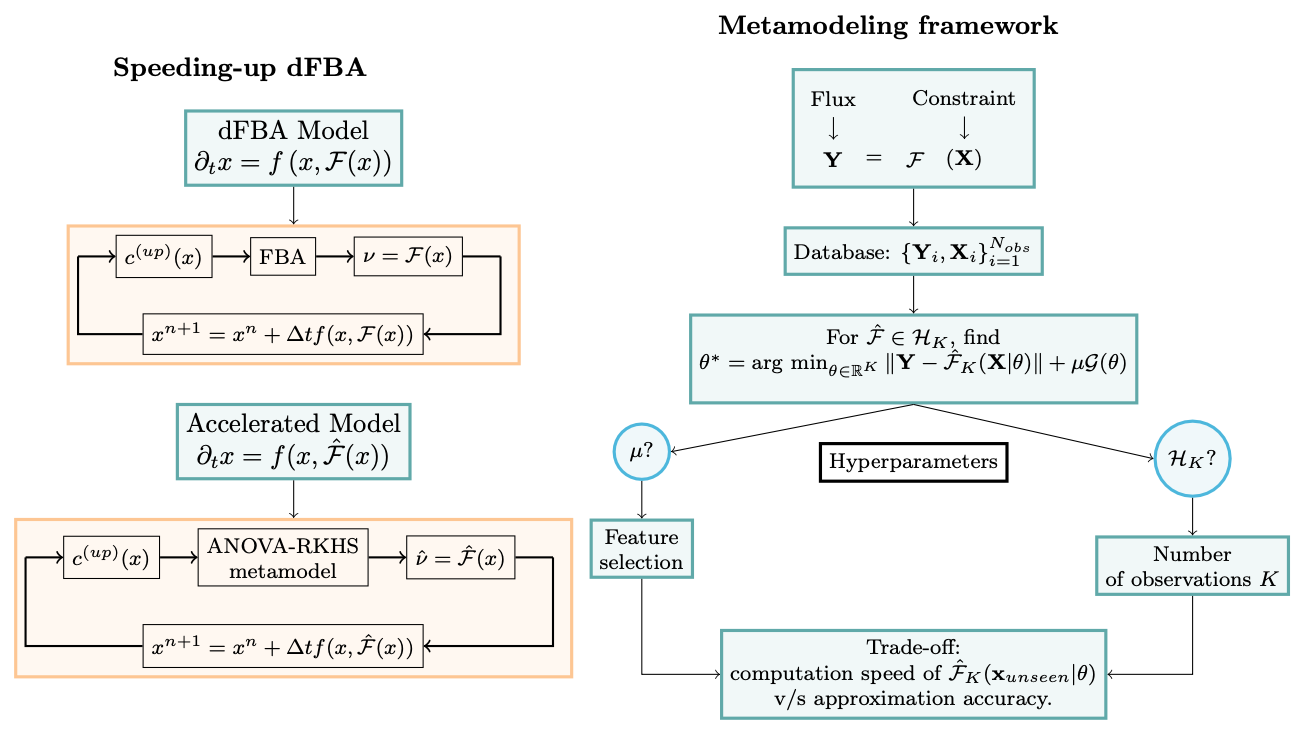
The figure contains two columns. On the left is a contrast of two iterative computations: one labeled “dFBA Model” with an FBA optimization step, one labeled “Accelerated Model” with a metamodel optimization step. On the right is a flowchart showing how hyperparameters are chosen, with a trade-off between computation speed and approximation accuracy.
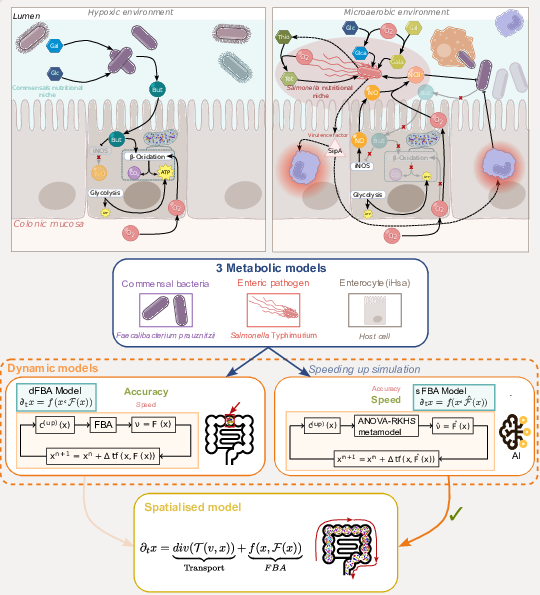
The upper part of the figure describes the biological system that is modelled: in absence of infection (upper left), and in infection conditions (upper right). The lower part of the figure illustrates the modelling procedure: three metabolic models are used to represent the three partners (human host, commensal bacteria and pathogen). Modelling the dynamics of the infection can be performed using dFBA or with a surrogate. Using the latter is more efficient computationally and permits building spatio-temporal models of the infection.
Another topic related to the PhD project of Maxime Lecomte consisted in integrating heterogenous omics data to build a dynamic model of cheese production. A preprint was submitted in 2023 22. It highlights a methodological approach for the construction and validation of dynamic metabolic models using dFBA. Using metabolic models for three bacterial species involved in cheese making, a careful curation and validation of metabolic functions was performed. Individual dynamics were calibrated using pure culture experimental data. The dynamics was then validated at the community level, illustrating the roles of each microbe in the production of flavour compounds.
8.2 Reasoning-based models of metabolism
Participants: Maxime Lecomte, David Sherman, Clémence Frioux, Chabname Ghassemi Nedjad, Léonard Brindel.
Over the past few years, we demonstrated how reasoning approaches can prove helpful for the analysis of microbial community metabolism through the Modeling of complementarity accross metabolic networks that permit the selection of minimal communities 45 and more generally screening metabolic potential in microbiomes 35.
These previous works illustrated the need for scalable methods in order to tackle large collections of genome, an objective that is hardly reachable with numerical models. Our recent work focuses on going further in characterizing microbial communities and in particular the interactions that occur among species and with their environment. We take advantage again of the expressivity of the logic paradigm of Answer Set Programming (ASP) as we provide a model of competitive and cooperative interactions among species in order to compare communities. As such, in the context of Maxime Lecomte's PhD, we developped scores assessing both competition and cooperation potentials based on the genome-scale metabolic networks of microbial species.
A second current axis of research relates to the PhD project of Chabname Ghassemi Nedjad. She develops an approach for the identification of seed metabolites in metabolic networks. The underlying biological challenge is to facilitate the culture of microbial dark matter, micro-organisms that cannot be cultured in controlled media experimentally. Usually, these organisms exhibit auxotrophies that prevent their growth on most nutritional conditions. We take advantage of the possibility to reconstruct metabolic networks from genomic information, therefby obtaining a blueprint of the metabolic potential of species. We apply a set of rules and constraints with the reasoning paradigm of ASP to these metabolic networks and provide sets of metabolites that would enable the activation of functions of interest in the corresponding species. Early results were presented by Chabname at a resercher summer school 23.
Our reasoning based models provide a lot of information regarding the role of each member of the microbial community, and mechanistic data on the concrete functions, genes and activity responsible for the predicted behaviour. As such, they can be very valuable. A remaining question though is the means to make all this information accessible and directly usable by the community of users who is not familiar with such modelling. To answer this question, Léonard Brindel was hired as an engineer in October 2023 and work on developing a platform associated to the sofware programme Metage2Metabo (M2M) 35. This platform will provide tools for analysing the wealth of data generated by M2M in light of additional metadata that users possess regarding their samples.
8.3 Metabolic Modeling of marine algae: methodological developments and applications
Participants: Clémence Frioux.
Brown algae, especially the species Ectocarpus siliculosus, are important models for deciphering the complex interactions within marine holobionts, with the goal of studying their metabolism together with the metabolism of the bacteria that inhabit their direct environment. Because performing wet lab experiments on such systems is technocally challenging, there is need for bioinformatic predictive methods for assessing the putative roles and interdependences between species. In parallel, addressing the difficulties brought by the study of these organisms is also a means to enhance and calibrate the tools devlopped in the team. Hence a fruitful collaboration for the past years has been developed with scientists from the Roscoff Biological station and the Inria project team Dyliss (Rennes).
The metabolism of algae is challenging to decipher, as it is for most eukaryotic species. A bottleneck resides in the difficulty of annotating their genomes, leading to manual annotations being of importance for these species. However, when aiming at comparing the metabolism of several organisms, it is preferable to annotate all of their genomes homogeneously, usually automatically, in order to prevent the propagation of biases during metabolic reconstruction. This entails ignoring the manual curation work performed on the genomes. AuCoMe is a pipeline aiming at preserving and propagating manual annotations as it permits working with heterogeneously-annotated genomes and efficiently compare their metabolic contents. The manuscript describing this pipeline was published in 2023 13. It was tested on several datasets of genomes, including algal ones, and demonstrated that it builds good quality metabolic networks as it corrects the inconsistencies brought by heterogenous annotation efforts.
8.4 Addressing the continuum of ecotypes in the gut using enterosignatures
Participants: Clémence Frioux.
The human gut microbiome generally hosts a few hundreds of microbial species, forming a dynamic but mostly stable ecosystem that can nevertheless deteriorate following perturbations. Being able to classify the state of the microbiome and identify dysbiotic conditions is of the highest interest for medical applications. Using ordination techniques or clustering approaches enables simplifying the composition of the gut microbiomes, ensuring discrete divisions and that a sample associates to one and only type 40. Such classification however lacks resolution and quantitative information regarding the proportional representation of microbial guilds in samples. To address these shortcomings, we proposed an ecologically informed decomposition of human gut microbiomes into five microbial signatures that we call 'enterosignatures' 15. The latter were obtained by applying non-negative matrix factorisation to the genus-level composition of more than 5,000 metagenomic samples.
We show that most samples of our dataset, that includes a large diversity of individuals from infants to elders, can be accurately described by a weighted combination of 2 or 3 enterosignatures. The relative proportion of each guild in the microbiome composition associates to metadata, for instance the enterosignature (ES) dominated by Firmicutes genera associates positively to health indicator, whereas we found associations between the Bacteroides-enriched ES and antibiotics consumption.
We demonstrated the generalisability of the model by applying the 5 ES to another dataset of European adults. We also assessed its relevance on an additional metacohort consisting of more than 1,100 samples from individuals with a non-western lifestyle. Results highlighted that the 5-ES model fits well to most samples, and evidenced a few cohorts with bad fitting scores whose original samples were not cold-stored, sugesting alterations in microbial community composition.
We showed that studying the samples that fit the least to the 5-ES model provides important information regarding putative perturbations of the gut microbiome. Atypical ES composition often associated to perturbed ecosystems such a antibiotic treatment or preterm birth. Altogether, we provide a simple and informative model to describe the composition of the human gut microbiome, that is generalisable to a wide range of individuals and permits highlighting perturbations of the ecosystem.
8.5 Functional profiling of the gut microbiota
Participants: Simon Labarthe.
With the emergence of metagenomic data, multiple links between the gut microbiome and the host health have been shown. Deciphering these complex interactions require evolved analysis methods focusing on the microbial ecosystem functions. Despite the fact that host or diet-derived fibres are the most abundant nutrients available in the gut, the presence of distinct functional traits regarding fibre and mucin hydrolysis, fermentation and hydrogenotrophic processes has never been investigated.
In 16, after manually selecting 91 KEGG orthologies and 33 glycoside hydrolases further aggregated in 101 functional descriptors representative of fibre and mucin degradation pathways in the gut microbiome, we used nonnegative matrix factorization to mine metagenomic datasets. Four distinct metabolic profiles were further identified on a training set of 1153 samples, thoroughly validated on a large database of 2571 unseen samples from 5 external metagenomic cohorts and confirmed with metatranscriptomic data. Profiles 1 and 2 are the main contributors to the fibre-degradation-related metagenome: they present contrasted involvement in fibre degradation and sugar metabolism and are differentially linked to dysbiosis, metabolic disease and inflammation. Profile 1 takes over Profile 2 in healthy samples, and unbalance of these profiles characterize dysbiotic samples. Furthermore, high fibre diet favours a healthy balance between Profiles 1 and Profile 2. Profile 3 takes over Profile 2 during Crohn's disease, inducing functional reorientations towards unusual metabolism such as fucose and H2S degradation or propionate, acetone and butanediol production. Profile 4 gathers under-represented functions, like methanogenesis. Two taxonomic makes up of the profiles were investigated, using either the covariation of 203 prevalent genomes or metagenomic species, both providing consistent results in line with their functional characteristics. This taxonomic characterization showed that Profiles 1 and 2 were respectively mainly composed of bacteria from the phyla Bacteroidetes and Firmicutes while Profile 3 is representative of Proteobacteria and Profile 4 of methanogens.
Integrating anaerobic microbiology knowledge with statistical learning can narrow down the metagenomic analysis to investigate functional profiles. Applying this approach to fibre degradation in the gut ended with 4 distinct functional profiles that can be easily monitored as markers of diet, dysbiosis, inflammation and disease.
8.6 Metagenomic assembly of complex microbial ecosystems
Participants: Nicolas Maurice, Léonard Brindel, Ariane Badoual, Franck Salin, Clémence Frioux.
The interest of Pleiade for the treatment of DNA sequences has renewed over the past few years with the development of a platform dedicated to the reconstruction of genomes from metagenomes, referred to as MAGs. This activity of the team is in line with the continuum of data from the sequences to the model. In 2023, Léonard Brindel worked, during his apprenticeship, on the validation of the platform initiated by Ariane Badoual.
During his intership co-supervised by Claire Lemaitre (Inria centre at the University of Rennes, Genscale team), Nicolas Maurice assessed the ability for existing metagenomic assemblers to accurately reconstruct genomes from increasingly complex communities. He developped a pipeline performing the comparison 24. This work was funded by the PEPR Agroecology and ICT. Nicolas Maurice was then hired as a PhD student in October 2023 and started a project aiming at developing new methods to facilitate the assembly of complex metagenomes such as those of the soil. He is co-supervised by Claire Lemaitre, Ricardo Vicedomini (Inria centre at the University of Rennes, Genscale team) and is hosted in the Genscale Inria team.
8.7 Statistical Modeling
Participants: Mohammed Anwar Abouabdallah, Alain Franc.
Mohamed-Anwar Abouabdallah defended his PhD thesis on Approche Tenseur-Train pour l'inférence dans les modèles à blocs stochastiques, application à la caractérisation de la biodiversité, accessible at theses.fr/2023BORD0023, on February 2nd, 2023. It presents a new approach for computing the marginals of a Weighted Stochastic Block Model from the Tensor-Train decomposition of the joint distribution. As TT decomposition provides separation of variables, computation of marginals follows swiftly. However, some numerical difficulties are met in the implementation, which have been addressed, and remain a challenging perspective. The PhD has been co-supervised by Nathalie Peurard (Inrae, MIAT, Toulouse), Olivier Coulaud (Inria BSO, Concace) and Alain Franc.
These methods are used for characterizing molecular biodiversity, building OTUs from a pairwise distance matrix using Stochastic Block Models (SBM). Building OTUs is traditionally seen as a form of unsupervised clustering. This work is done in collaboration with the MIAT INRAE research unit in Toulouse and HiePACS. It represents a connection between metabarcoding and statistical modeling, a topic which deserves investigation. Previously, in 31, we studied whether morphological-based and molecular-based approaches are in agreement, and provided evidence that yes, automatic clustering and group identification can be done reliably using barcoding. Using Aggregative Hierarchical Clustering and Stochastic Block Models, we found that the agreement between morphological-based and molecular-based classifications ranges in most cases from good to very good at taxonomic levels above species (figure 3).
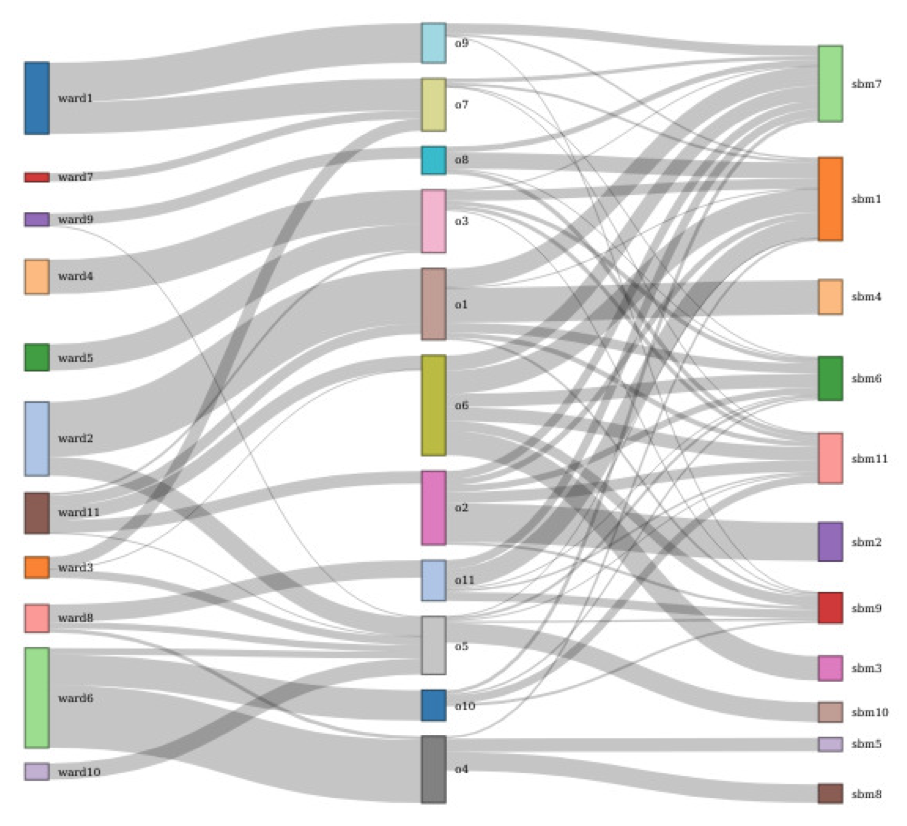
The figure contains three columns comprised of vertically-oriented rectangles of different sizes and colors. Vertical intervals of the colored rectangles in adjacent columns are linked by gray horizontal paths, curved so that the paths are perpendicular to the rectangles, but change vertical position to establish the correspondence between the intervals of the rectangles. A single rectangle may thus be linked to several rectangles in the adjacent column. Overall, the intervals cover most of the extent of the rectangles. To the left, most rectangles in the middle column map to just one, or to a small number, of rectangles in the left column. To the right, the relations are more complex: rectangles in the middle column often map to several rectangles in the right column, and, conversely, rectangles in the right column may map to several rectangles in the middle column.
8.8 Metabarcoding and Taxonomic Diversity
Participants: Jean-Marc Frigerio, Alain Franc.
Metabarcoding is a series of technical procedures to build molecular based inventories from large datasets of amplicons. The underlying information needs to be compacted without losing its information content before it can be further processed with domain-specific tools. This links metabarcoding tools to dimension reduction techniques, which is an important topic in Pleiade.
Alain Franc and Jean-Marc Frigerio have continued their efforts to complete the yapotu software (§7.1.6), which makes the main stages of barcoding and metabarcoding data analysis accessible. The main progress in 2023 has been, beyond the development of some methods, the development of a Domain Specific Language, in Python, called yapsh, that makes the execution of instructions more automatic and comfortable for the user. The project, still in development, is accessible publicly on the Inria Gitlab.
The work on the evaluation of the intrinsic quality of OTUs produced in metabarcoding (Marie-Josée Cros, Jean-Marc Frigerio, Nathalie Peyrard and Alain Franc,Simple approaches for evaluation of OTU quality based on dissimilarity arrays) , has been achieved by submitting a paper to the Journal MBMG, currently in revision. The preprint of the manuscript is available at 41. As a companion to this work is available in a Git project on the public “Forge MIA” of the Inrae. It contains the documentation for the programs implementing the methods, as well as a toy data set with a tutorial on how to use it.
8.9 Dimension Reduction
Participants: Alain Franc.
Version 3 of Alain Franc's comprehensive report on Linear Dimensionality Reduction, Research Report 9488, Inria Bordeaux Sud-Ouest. 2023, 99 pages, is available at 21 and arXiv 2209.13597. The main changes from version 2 are:
- the presentation of Quadratic embedding as an alternative to MDS when some eigenvalues of the Gram matrix are negative
- the redaction of three appendices, on on preliminaries in linear algebra, one on quadrative forms (for quadratuic embedding), and one on a reason why random projection works.
9 Bilateral contracts and grants with industry
In 2023 Pleiade's impact on industry was channeled through agroecological project MISTIC (§10.3.1) and did not require bilateral projects.
10 Partnerships and cooperations
10.1 International initiatives
10.1.1 Associate Teams in the framework of an Inria International Lab or in the framework of an Inria International Program
Clémence Frioux co-leads the Inria associated team Symbiodiversity with the University of Santiago de Chile.
10.2 International research visitors
10.2.1 Visits of international scientists
Other international visits to the team
Constanza Andreani
-
Status
Engineer
-
Institution of origin:
Center for Genome Regulation, Santiago de Chile
-
Country:
Chile
-
Dates:
2023-10 / 2023-11
-
Context of the visit:
Symbiodiversity
-
Mobility program/type of mobility:
(sabbatical, internship, research stay, lecture…)
Connor Tifany
-
Status
PhD
-
Institution of origin:
University of California, Davis
-
Country:
USA
-
Dates:
2023-06-12 / 2023-06-15
-
Context of the visit:
Collaboration UC Davis
-
Mobility program/type of mobility:
research stay, lecture
10.2.2 Visits to international teams
Frioux Clémence
-
Visited institution:
University of Louvain
-
Country:
Belgium
-
Dates:
2023-07-03 / 2023-07-04
-
Context of the visit:
Invited seminar
-
Mobility program/type of mobility:
lecture
10.3 National initiatives
10.3.1 MISTIC (PEPR Agroecology and ICT)
Participants: David Sherman, Clémence Frioux, Simon Labarthe, Alain Franc, Jean-Marc Frigerio.
MISTIC, Microbial communities and ICT, has been selected as a five-year flagship project in the PEPR Agroecology and ICT program of the French Government. MISTIC will develop methodological tools for defining spatio-temporal models of microbial community dynamics in the phyllosphere and rhizosphere of crop plants, with the goal of creating new understanding of the role of these communities in plant adaptation to environmental stresses, including climate change. MISTIC is a partnership between seven Inria and INRAE teams in Bordeaux, Rennes, and Sophia Antipolis. The project formally began in November 2022.
10.3.2 VITAe, Pherosensor (PPR Cultiver et proteger autrement)
Participants: Simon Labarthe.
Pleiade participates to two projects of the PPR CPA, dedicated to research towards an agriculture without pesticide. Pléiade co-leads a work package of the VITAe project, taking in charge modeling tasks to analyse culturomics data in order to identify antagonist micro-organisms against powdery mildew in grapewine. Pléiade leads a work package of the Pherosensor project, dedicated to the design of new sensors of pheromone. The main task of the team is to solve an inverse problem on a PDE model of pheromone propagation to track back the pheromone emitters.
10.3.3 Holovini (Holoflux metaprogram)
Participants: Simon Labarthe, Clémence Frioux.
Pleiade is involved in the Holovini flagship project of the Holoflux metaprogram. Holovini studies the berry microbiome of grapewine, focusing on the microbial flux involved in the assembly of the berry microbiome. Pléiade takes in charge the analysis of metagenomics data and the co-lead of a modeling workpackage.
10.3.4 REBON (ANR)
Participants: Clémence Frioux.
REBON, piloted by Joachim Niehren, will abstract reaction networks to boolean networks with the goal of improving inference and control in systems biology.
10.3.5 Artemis (Digit-bio metagragram)
Participants: Simon Labarthe, Clémence Frioux, David Sherman.
Pleiade pilots the Artemis pre-project funded by the Digit-bio metaprogram, aimed at developing methodologies for defining digital twins in microbial ecology.
10.3.6 Agence Française pour la Biodiversité
Participants: Alain Franc, Jean-Marc Frigerio.
The AFB is a public law agency of the French Ministry of Ecology that supports public policy in the domains of knowledge, preservation, management, and restoration of biodiversity in terrestrial, aquatic, and marine environments. Pleiade is a partner in two AFB projects developed with the former ONEMA: one funded by ONEMA, the second by labex COTE, where BioGeCo/Pleiade is responsible for data analysis, with implementaton of the tools recently developed for scaling MDS. Calculations have been made on CURTA at MCIA and PlaFRIM at INRIA.
10.4 Regional initiatives
10.4.1 Poppy Rosa ProtoImpact (CRNA)
Participants: David Sherman.
Poppy Rosa ProtoImpact is an educational robots project financed by the Conseil Régional de la Nouvelle Aquitaine (CRNA) and coordinated by the Ligue de l'Enseignement de la Gironde, with Poppy Station and Inria as partners. Poppy Rosa is prototyping a new way of teaching concepts of algorithmics and artificial intelligence to young students, with the goal of training tomorrow's citizens. Our deployment specifically targets rural schools across the Neo-Aquitaine region.
The first version of the Poppy Rosa hardware and software platform was presented to collaborators from Biblio.Gironde and is being deployed in libraries and schools.
Poppy Rosa uses parts designed and manufactured by David Sherman in the EirLab (ENSEIRB engineering school, Talence) and eurêkafab (Communauté de Communes de Montesquieu, Martillac) fab labs.
10.4.2 UCIA, Usages et connaissances de l'intelligence artificielle
Participants: David Sherman.
The Ligue de l'Enseignement, fédération de Gironde, has developed the outreach project Usages et connaissances de l'intelligence artificielle (UCIA) led by Coline Delbos. Organized in three one-hour modules, the activity aims to provide young adults with a basic understanding of AI and our choices concerning it in our day-to-day lives.
11 Dissemination
11.1 Promoting scientific activities
11.1.1 Journal
Member of the editorial boards
- BMC Evolutionary Biology – Alain Franc
Reviewer - reviewing activities
- Cell Report Medicine – Clémence Frioux
- mSystems – Clémence Frioux , Simon Labarthe
- Nature Communications – Clémence Frioux
- Peer Community In (PCI) Math Comp Biol – Clémence Frioux
- NPJ biofilms – Simon Labarthe
- Journal of biological systems – Simon Labarthe
11.1.2 Invited talks
- EBAME workshop, Brest (France) – Seminar – Addressing the continuum of ecotypes in the gut microbiome with enterosginatures – Clémence Frioux
- Plant Health Institute Montpellier, France – Seminar – In silico exploration of microbial ecosystems with metabolic models – Clémence Frioux
- KU Leuven, Belgium – Seminar – Exploration of microbial ecosystems: from compositional patterns to metabolic models – Clémence Frioux
- INRAe Metagenopolis, Jouy en Josas, France – Seminar – Microbial communities: from data to models [Clémence Frioux & Simon Labarthe]
- INRAe EGFV unit, Bordeaux – Seminar – Microbial communities: from data to models – Clémence Frioux , Simon Labarthe
- INRAe Apero Vitae, Bordeaux – Seminar – Microbial communities: from data to models – Clémence Frioux , Simon Labarthe
- Ferment'IA, Saclay-Supelec (France) – workshop – Vers un jumeau numérique du métabolisme bactérien pendant la production de fromage – Simon Labarthe
- INRAe Micalis, Jouy-en-Josas, France – Mini-symposia – Modeling defined community of the gut – Simon Labarthe
- Journée IFPEN–Inria, Rueil Malmaison (France) – workshop – Stockage CO : couplage avec la biologie – David Sherman
- Journées INRAE–Inria, Champenoux (France) – workshop – Computational Models of Crop Plant Biodiversity – David Sherman
- Conseil d'administration du département Génétique Animale, Paris (France) – invited talk – MISTIC – Microbial communities and ICT – David Sherman
- Journées Agroécologie BIOSENA, Bordeaux (France) – workshop – Microbiomes de plantes cultivées – David Sherman
- Inauguration PEPR Agroecology, Paris (France) – workshop – Computational Models of Crop Plant Biodiversity – David Sherman
11.1.3 Leadership within the scientific community
- David Sherman is on the steering committee of Biosena, a regional research network of the New Aquitaine region dedicated to Biodiversity and Ecosystemic Services. Biosena associates actors from the academic and socio-economic sectors, with the goal of contributing to the understanding and preservation of biodiversity and to the improvement of ecosystemic services. Biosena contributes to this goal through research, knowledge dissemination, outreach, and skill transfer in the form of Research Action, in keeping with the recommendations of Ecobiose.
- David Sherman is member of the board (membre du Conseil d'administration) and secretary of the Mobsya Association, Lausanne. Mobsya develops and commercializes the Thymio educational robot, geared towards K-12.
- David Sherman is member of the board (membre du Conseil d'Administration) and lead advisor for software of the Poppy Station Association. Poppy Station develops open-hardware open-source humanoid robots for research and education.
11.1.4 Scientific expertise
Recruitment committees
- Junior researcher selection committee of the Plant Health Department of INRAe – Clémence Frioux
11.1.5 Research administration
Local responsabilities
- Co-creation of and participation in a gender equality and diversity working group in the Inria Centre at the University of Bordeaux – Clémence Frioux
- Resource person for the 3D printing and electronics workshop – David Sherman
- Workplace first-aider (SST) – David Sherman
National responsabilities
- Project coordinator, MISTIC (§10.3.1) – David Sherman
- Participation to the Inria national committee for equality and inclusion – Clémence Frioux
- Participation to the Holoflux INRAe metaprogram steering committee.
- Participation to the CSS (commission scientifique spécialisée) MISTI of INRAe
11.2 Teaching - Supervision - Juries
11.2.1 Teaching
- Master – ENSTBB Bordeaux INP - Bioinformatics – Clémence Frioux
- Master – ENSEIRB Bordeaux INP - Research algorithms – Clémence Frioux
- EBAME workshop, Brest (France) – In silico exploration of metabolism in microbial ecosystems: from the metabolic network to the model – Clémence Frioux
11.2.2 Supervision
- PhD of Maxime Lecomte (2020-2024) - Approches hybrides en modélisation logique et numérique du métabolisme des écosystèmes microbiens - David Sherman (Director), Hélène Falentin (director, INRAE STLO), Clémence Frioux (advisor)
- PhD of Chabname Ghassemi Nedjad (2022-2025) - Combinatorial optimisation problems for reverse ecology - Clémence Frioux (co-director), Loïc Paulevé (CNRS, LaBRI, co-director).
- PhD of Nicolas Maurice (2023-2026) (Genscale, Inria centre at the university of Rennes) - Algorithmique des séquences pour la reconstruction de génomes à partir de données métagénomiques complexes - Claire Lemaitre (Inria, director), Ricardo Vicedomini (CNRS, co-director), Clémence Frioux (co-advisor).
- PhD of Amandine Paulay (2020-2024) (Micalis, Inrae Jouy en Josas) - Modélisation de la dégradation des protéines alimentaires par le microbiote intestinal humain - Emmanuelle Maguin (Director, Inrae, Micalis), Beatrice Laroche (co-Director, INRAe, MaIAGE), Simon Labarthe (advisor, INRAe-Inria, Biogeco-Pléiade), Ghjuvan Grimaud (supervisor, Biomathematica)
- Postdoc of Pablo Ugalde Salas - co-supervised by Clémence Frioux and Simon Labarthe
- Postdoc of Thibault Malou (INRAe, Maiage) - Simon Labarthe
- Master's internship of Rafael Kaempfer Danin - co-supervised by Simon Labarthe and Pablo Ugalde Salas
- Master's apprenticeship of Léonard Brindel - co-supervised by Clémence Frioux and Franck Salin
- Master's intership of Nicolas Maurice - co-supervised by Claire Lemaitre (Inria, Genscale, Rennes) and Clémence Frioux
11.2.3 Juries
PhD defense juries
- Clémence Joseph (KU Leuven, Belgium) – Clémence Frioux – examiner
- Marie Burel (Univ. Evry, France) – Clémence Frioux – examiner
PhD thesis committes
- Morhane Roge (Grenoble) – Clémence Frioux
- Clément Gavoille (Bordeaux) – David Sherman
11.3 Popularization
11.3.1 Articles and contents
- Clémence Frioux and Simon Labarthe wrote a popularisation article for the Interstices website 28.
11.3.2 Education
- Clémence Frioux taught two small workshops during the "MIMM, moi informaticienne, moi mathématicienne" 2023 week, a free internship at the University of Bordeaux for young girls in 9th and 10th grade in order to encourage them to choose mathematics and computer science, allows them to discover training, research and jobs in these two disciplines.
11.3.3 Interventions
- Chiche ! Un ou une scientifique, une classe – Clémence Frioux 5 classes, David Sherman 3 classes
- Science festival (Fête de la Science) at Inria Bordeaux – Clémence Frioux 2 presentations, David Sherman 3 activities Qui se ressemble s'assemble
- Journées emploi en maths (Université de Bordeaux) – Simon Labarthe
- Numérique Éthique Tour (Cabane à Projets, Créon) – Usages et connaissances de l'intelligence artificielle – David Sherman
12 Scientific production
12.1 Major publications
- 1 articleA Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum..Nature Communications52014, 4044HALDOIback to text
- 2 articleModeling acclimatization by hybrid systems: Condition changes alter biological system behavior models.BioSystems121June 2014, 43-53HALDOI
- 3 articleMetagenomic assessment of the global distribution of bacteria and fungi.Environmental MicrobiologyNovember 2020HALDOI
- 4 articleMetage2Metabo, microbiota-scale metabolic complementarity for the identification of key species.eLife9December 2020HALDOIback to text
- 5 articleMetabolic Complementarity Between a Brown Alga and Associated Cultivable Bacteria Provide Indications of Beneficial Interactions.Frontiers in Marine Science7February 2020, 1-11HALDOI
- 6 articleThe genome of Ectocarpus subulatus – A highly stress-tolerant brown alga.Marine Genomics52January 2020, 100740HALDOI
- 7 articleEnterosignatures define common bacterial guilds in the human gut microbiome.Cell Host & MicrobeJune 2023HALDOI
- 8 articleUsing automated reasoning to explore the metabolism of unconventional organisms: a first step to explore host–microbial interactions.Biochemical Society Transactions483May 2020, 901-913HALDOIback to text
- 9 articleFrom bag-of-genes to bag-of-genomes: metabolic modelling of communities in the era of metagenome-assembled genomes.Computational and Structural Biotechnology JournalJune 2020HALDOI
- 10 incollectionWhy We Need Sustainable Networks Bridging Countries, Disciplines, Cultures and Generations for Aquatic Biomonitoring 2.0: A Perspective Derived From the DNAqua-Net COST Action.Next Generation Biomonitoring: Part 158Elsevier2018, 63-99HAL
- 11 articleExact or approximate inference in graphical models: why the choice is dictated by the treewidth, and how variable elimination can be exploited.Australian and New Zealand Journal of Statistics612to appearJune 2019, 89-133HALDOI
- 12 articleGenolevures: protein families and synteny among complete hemiascomycetous yeast proteomes and genomes..Nucleic Acids Research372009, D550-D554HALDOIback to text
12.2 Publications of the year
International journals
International peer-reviewed conferences
Edition (books, proceedings, special issue of a journal)
Reports & preprints
Other scientific publications
12.3 Other
Scientific popularization
12.4 Cited publications
- 30 articleGénolevures – a novel approach to ‘evolutionary genomics’.FEBS Letters (complete issue)48712000, URL: https://febs.onlinelibrary.wiley.com/toc/18733468/2000/487/1back to text
- 31 articleDoes clustering of DNA barcodes agree with botanical classification directly at high taxonomic levels? Trees in French Guiana as a case study.Molecular Ecology Resources2022HALDOIback to textback to text
- 32 articleTraceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models.PLOS Computational Biology1452018, e1006146DOIback to text
- 33 articleRADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling.Mol. Ecol.22112013, 3179--90back to text
- 34 articleThe Euclidean Distance Matrix Completion Problem.SIAM J. Matrix Anal. App.1621995, 646-654back to text
- 35 articleMetage2Metabo, microbiota-scale metabolic complementarity for the identication of key species.eLife92020, e61968DOIback to textback to text
- 36 articleExploring the Bacterial Impact on Cholesterol Cycle: A Numerical Study.Frontiers in Microbiology112020, 1121HALDOIback to text
- 37 articleExact Matrix Completion via Convex Optimization.Found. Comput. Math.92009, 717-772back to text
- 38 bookParasitism: The Ecology and Evolution of Intimate Interactions.University of Chicago Press2001back to text
- 39 articleComparative genomics of protoploid Saccharomycetaceae.Genome Research192009, 1696-1709URL: http://hal.inria.fr/inria-00407511/en/DOIback to text
- 40 articleEnterotypes in the landscape of gut microbial community composition.Nature Microbiology312018, 8--16DOIback to text
- 41 unpublishedSimple approaches for evaluation of OTU quality based on dissimilarity arrays.October 2022, working paper or preprintHALback to text
- 42 articleGenome evolution in yeasts.Nature4302004, 35-44URL: http://www.nature.com/nature/journal/v430/n6995/abs/nature02579.htmlDOIback to text
- 43 articleStructural analysis of expanding metabolic networks..Genome informatics. International Conference on Genome Informatics1512004, 35--45back to text
- 44 articleNonlinear mapping and distance geometry.Optimization Letters142May 2019, 453-467HALDOIback to text
- 45 articleScalable and exhaustive screening of metabolic functions carried out by microbial consortia.Bioinformatics34172018, i934--i943DOIback to textback to textback to text
- 46 articleHybrid metabolic network completion.Theory and Practice of Logic Programming1912019, 83--108DOIback to text
- 47 articleReference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap.PLoS Genetic94e10034572013back to textback to text
- 48 articleClingo = ASP + Control: Preliminary Report.CoRRabs/1405.36942014back to text
- 49 inproceedingsAdvances in gringo Series 3.LPNMR6645Lecture Notes in Computer ScienceSpringer2011, 345---351back to text
- 50 articleA mathematical model to investigate the key drivers of the biogeography of the colon microbiota..Journal of Theoretical Biology46272019, 552-581HALDOIback to text
- 51 articleEuclidean Distance Geometry and Applications.SIAM review56(1)2014, 3-69back to text
- 52 articleEstimation of Nucleotide Diversity, Disequilibrium Coefficients, and Mutation Rates from High-Coverage Genome-Sequencing Projects.Mol. Biol. Evol.25112008, 2409--19back to text
- 53 articleWhat is flux balance analysis?Nature Biotechnology2832010, 245--248DOIback to text
- 54 articleMeneco, a Topology-Based Gap-Filling Tool Applicable to Degraded Genome-Wide Metabolic Networks.PLOS Computational Biology1312017, e1005276DOIback to text
- 55 articleInferring characteristics of bacterial swimming in biofilm matrix from time-lapse confocal laser scanning microscopy.eLife11June 2022HALDOIback to text
- 56 articleA comprehensive framework for global patterns in biodiversity.Ecology Letters712004, 1--15URL: http://dx.doi.org/10.1046/j.1461-0248.2003.00554.xDOIback to text
- 57 articleGenolevures complete genomes provide data and tools for comparative genomics of hemiascomycetous yeasts..Nucleic Acids Research34Database issueJanuary 2006, D432-5HALDOIback to text
- 58 articleGénolevures: protein families and synteny among complete hemiascomycetous yeast proteomes and genomes.Nucleic Acids Research37suppl 12009, D550-D554URL: http://nar.oxfordjournals.org/content/37/suppl_1/D550.abstractDOIback to textback to text
- 59 articleEcology, evolution, and epidemiology of zoonotic and vector-borne infectious diseases in French Guiana: Transdisciplinarity does matter to tackle new emerging threats.Infection, Genetics and Evolution93September 2021, 104916HALDOIback to text
- 60 articleThe influence of habitat on viral diversity in neotropical rodent hosts.Viruses139August 2021, 1-29HALDOIback to text