Section: Overall Objectives
Presentation
The team Bonsai has been re-created on January 1, 2011, and is an evolution of the INRIA-LIFL team Sequoia, which was created in 2007. The scientific focus of Bonsai is still very much the same as the one of Sequoia. We work in computational biology, and more specifically o n algorithms for biological sequences analysis. Several topics of Bonsai were already present in Sequoia: Noncoding RNA analysis and non ribosomal peptide synthesis. We also work on further lines of research: Algorithms for Next Generation Sequencing and comparison of sequences at genome scale taking into account rearrangements. These lines of research find their source in the development of new sequencing technologies and the increasing availability of complete genome sequence data. They are supported by strategical collaborations, and they also reinforce the expertise of the team in sequence analysis and genome annotation. The main goal of Bonsai is to define appropriate combinatorial models and efficient algorithms for large-scale sequence analysis in molecular biology.
From a historical perspective, research in bioinformatics started with string algorithms designed for the comparison of sequences. Bioinformatics became then more diversified, accompanying the emergence of new high-throughput technologies. By analogy to the living cell itself, bioinformatics is now composed of a variety of dynamically interacting components forming a large network of knowledge: systems biology, proteomics, text mining, phylogeny, structural genomics, etc. Sequence analysis remains a central node in this interconnected network, and it is the heart of the Bonsai team. It is a common knowledge nowadays that the amount of sequence data available in public databanks grows at an exponential pace. Conventional DNA sequencing technologies developed in the 70's already permitted the completion of hundreds of genome projects that range from bacteria to complex vertebrates. This phenomenon is dramatically amplified by the recent advent of Next Generation Sequencing. From a computational point of view, NGS gives rise to many new challenging problems in computational biology.
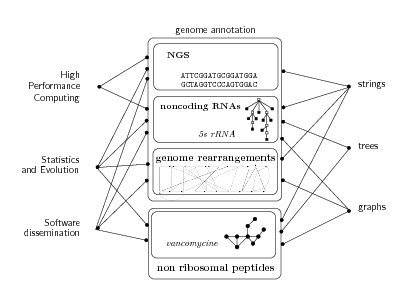
The completion of sequencing projects in the past few years also teaches us that the functioning of the genome is more complex than expected, which makes genomic sequence annotation a difficult task. Originally, genome annotation was mostly driven by protein-coding gene prediction. It is now widely recognized that deciphering gene regulation is an essential task to understand the function of a protein. Noncoding RNA genes also play a key role in many cellular processes. Besides proteins encoded in the genome, there exist other, more specific, processes such as nonribosomal peptide synthesis that produces small peptides not going through the central dogma. At a higher level, genome organization and rearrangements are also a source of complexity and have a high impact on the course of evolution.
All above-mentioned biological phenomena together with big volumes of new sequence data and new hardware provide a number of new challenges to bioinformatics, both on modeling the underlying biological mechanisms and on efficiently treating the data. Bonsai is a fully interdisciplinary project whose scientific core is the design of efficient algorithms for the analysis of biological macromolecules. We focus on four main themes: high-throughput sequence analysis, noncoding RNAs, nonribosomal peptides, and genome rearrangements. See Figure 1 for an illustration. Most of research projects are carried out in collaboration with biologists. A special attention is given to the development of robust software, its validation on biological data and its availability from the software platform of the team: http://bioinfo.lifl.fr/ .